Vasculitis
1.Introducción
Las vasculitis comprenden un grupo heterogéneo de entidades relativamente frecuentes (suponen 1 de cada 300 ingresos en un hospital universitario) de etiologías y manifestaciones diversas, que se caracterizan por la inflamación de los vasos sanguíneos, arterias, venas o ambos, lo cual compromete su función con el desarrollo de isquemia y necrosis.
La inflamación vascular puede acompañarse de sintomatología general (fiebre, astenia, afectación del estado general) y/o el desarrollo de manifestaciones locales orgánicas dependientes del órgano afecto por la vasculitis (afectación cutánea, síntomas neurológicos, dolor abdominal, compromiso renal, etc.). La piel y el tejido subcutáneo se afectan frecuentemente en las vasculitis. Esta alta frecuencia de afectación es probablemente debida a diversos factores incluyendo el gran número de vasos dermicos, la exposición al frio y la presencia de fenómenos de estasis vascular que favorecerían el desarrollo de vasculitis con afectación cutánea. Las vasculitis pueden manifestarse a nivel cutáneo de diversas formas, desde cambios en la coloración, edema, púrpura, equímosis y necrosis (úlceras), siendo la manifestación más frecuente el desarrollo de púrpura palpable en extremidades inferiores. El desarrollo de una púrpura palpable puede ser la manifestación de una vasculitis benigna de corta duración, puramente cutánea a una manifestación cutánea de una vasculitis sistémica que puede acompañarse de afectación orgánica con compromiso vital. La elevada frecuencia de manifestaciones cutáneas de las vasculitis hace que el reconocimiento de estas lesiones, sea importante para el diagnóstico de las mismas.
2.Patogénesis
El diagnóstico y la clasificación de las vasculitis se basa especialmente en los mecanismos patogénicos que las producen. Se pueden clasificar las vasculitis en base a los mecanismos de producción que incluyen: la infección directa de los vasos, mecanismos inmunes y vasculitis de causa desconocida (Tabla 1). La mayor parte de vasculitis pueden catalogarse dentro de las causas infecciosas o en las inmunológicas. Dado que el tratamiento de las vasculitis infecciosas es radicalmente diferente del de las vasculitis mediadas por daño inmunológico, es importante realizar la distinción entre las dos formas de vasculitis en las fases iniciales de la valoración de estos enfermos. Es importante descartar una causa infecciosa antes de instaurar un tratamiento inmunosupresor. La mayoría de las vasculitis están mediadas por mecanismos inmunes, y se clasifican según los 4 tipos de reacción de hipersensibilidad de Gell y Coombs:
Tabla 1 Mecanismos patogénicos de las vasculitis
|
|
- Tipo I (vasculitis alérgica o anafiláctica: incluye las vasculitis asociadas a estados atópicos, urticaria vasculitis y síndrome de Churg-Strauss. Se caracterizan por la presencia de niveles séricos y tisulares de IgE elevados. En la fase vasculítica se caracteriza por infiltración angiocéntrica de los vasos por eosinófilos.
- Tipo II (citotóxica o citolítica):
- vasculitis mediadas por ANCA (granulomatosis de Wegener, poliangeitis microscópica y síndrome de Churg-strauss). Los ANCA (anticuerpos anticitoplasma de neutrófilos) son capaces de activar los neutrófilos y las células endoteliales, así como inducir la apoptosis acelerada de los neutrófilos.
- Anticuerpos anti-células endoteliales endoteliales (AECA). Los AECA pueden causar vascultis por daño directo o por activación del complemento, están involucrados en la enfermedad de Behçet y la enfermedad de Takayasu y tienen especificidad por diferentes regiones vasculares, afectando a vasos de pequeño tamaño en la enfermedad de Behçet y vasos de gran tamaño en la enfermedad de Takayasu.
- Tipo III (mediada por inmunocomplejos): el depósito de inmunocomplejos da lugar a la activación del complemento y liberación de los componentes C3 y C5, que producen quimiotáxis de neutrófilos y liberación de enzimas proteolíticas que dañan la pared vascular. Es el grupo más amplio de vasculitis entre las que encontramos la vasculitis leucocitoclástica cutánea, el síndrome de Schonlein-Henoch y poliarteritis nodosa, entre otras formas.
- Tipo IV (citotóxica) vasculitis mediada por linfocitos T: en este grupo se incluyen aquellas vasculitis granulomatosas que se caracterizan por la presencia en la pared de los vasos de infiltrados inducidos por linfocitos T, especialmente Th1, que serían responsables por medio de la producción de interferon-γ, de la acumulación de macrófagos que fagocitarían las fibras elásticas. En este grupo de vasculitis se encuentra la arteritis de la temporal.
Las vasculitis son un grupo de síndromes heterogéneo que sin embargo comparten varias manifestaciones entre ellas. No existe un esquema de clasificación ideal de las vasculitis. Se pueden clasificar en relación a si
- son primarias o secundarias (tabla 2),
- en relación al tamaño del vaso afecto y las características histológicas encontradas en la histología (tabla 3 ) o
- en base a los mecanismos patogénicos (tabla 4).
4.Manifestaciones cutáneas de vasculitis
4.1. Clinica: Las manifestaciones clínicas cutáneas de las vasculitis en la piel orientan hacia el diagnóstico de vasculitis, pero no son especificas de ninguna entidad específica. Las lesiones cutáneas son útiles como signo diagnóstico y su estudio histológico y mediante inmunofluorescencia directa sirven para obtener confirmación diagnóstica de vasculitis. Las manifestaciones más características son el desarrollo de púrpura palpable y de nódulos, pero pueden observarse otras manifestaciones cutáneas tales como petequias, equimosis, máculas eritematosas, lesiones de urticaria, livedo reticularis, necrosis, ulceras, vesículas, pústulas, ampollas, lesiones a tipo pioderma gangrenoso, lesiones tipo eritema nodoso y lesiones a tipo síndrome de Sweet. El predominio de una lesión clínica u otra vendrá determinado por la localización del vaso afecto y por las características del proceso inflamatorio.
Las vasculitis con afectación de vasos de pequeño calibre en la piel se manifiestan principalmente por la púrpura, que con frecuencia es palpable y afecta principalmente a extremidades inferiores (debido a la presión hidrostática). La púrpura suele desarrollarse en brotes sucesivos, inicialmente son máculas de coloración rojiza, que evolucionan hacia placas y pápulas, que pueden ser desde unos milímetros hasta varios centímetros de diámetro. Las lesiones más grandes son mas equimóticas que purpúricas. El color puede evolucionar desde el rojo-purpúrico hasta parduzco, en relación a la evolución de la degradación de la sangre extravasada. En algunos pacientes, especialmente aquellos en los que la vasculitis mediada por inmunocomplejos se acompaña de una gran activación de complemento pueden dar lugar a focos de edema cutáneo que se manifiesta clínicamente por brotes de urticaria, que generalmente dura mas de 24 horas y evoluciona hacia lesiones purpúricas. Los nódulos suelen ser calientes, tumefactos y rojos y pueden estar rodeados por lesiones de livedo reticulares. Los nódulos cutáneos se observan en las vasculitis que afectan a vasos de mayor calibre, como la poliarteritis nodosa - cutánea o sistémica-, la granulomatosis de Wegener, el síndrome de Churg-Strauss y la arteritis de células gigantes.
Tabla 6 Manifestaciones cutáneas de las vasculitis
4.2 Histología: El hallazgo histológico más frecuente en las vasculitis es el de es una vasculitis neutrofílica leucocitoclastica , que se caracteriza por la presencia de infiltrado inflamatorio afectando a la pared vascular con presencia de edema endotelial, infiltrado inflamatorio con predominio de polimorfonucleares neutrófilos, leucocitoclasia (degranulación y fragmentación de los polimorfonucleares dando lugar al polvo nuclear) hemorragia y trombosis. El desarrollo de nódulos cutáneos en pacientes con vasculitis pueden observarse en los casos en que existe un inflamación cutánea o subcutánea centrada en un vaso. La realización de estudios de inmunofluorescencia directa son útiles en el diagnóstico de vasculitis ya que nos permiten la demostración de inmunoclomplejos depositados en los vasos dérmicos y saber por que inmunoglobulina están constituidos (IgG, IgM, IgA, C3 y/o fibrinogeno).
5.Síndromes vasculíticos
Las vasculitis que nos van a interesar más son las vasculitis sistémicas primarias producidas por trastornos inmunes como la vasculitis o angeitis leucocitoclástica cutánea, las vasculitis sistemicas primarias asociadas a ANCAS incluyendo la granulomatosis de Wegener, la granulomatosis de Churg-Straus y la poliangeitis microscopica, asi como las vasculitis sistemicas con afectación de vasos de gran tamaño como arteritis temporal y la poliarteritis nodosa.
5.1.Vasculitis por hipersensibilidad o vasculitis leucocitoclastica cutánea
El término de vasculitis leucocitoclastica o necrotizante cutánea engloba un grupo amplio y heterogéneo de síndromes que se caracterizan por la inflamación, mediada por inmunocomplejos, de vasos capilares, vénulas y ocasionalmente arteriolas cutáneas, con cambios histológicos que se describen bajo el término de vasculitis leucocitoclástica (edema endotelial, infiltración por polimorfonucleares, cariorexis, hemorragia y trombosis).Incidencia: La vasculitis por hipersensibilidad es la forma más frecuente de vasculitis, representando entre el 17-29% de los casos de vasculitis. Puede afectar a cualquier edad, el 10% de los afectos son niños. Clínica: Las manifestaciones clínicas más frecuentes son dermatológicas. Más del 95% de los pacientes desarrollan púrpura palpable. Otras manifestaciones clínicas incluyen urticaria, eritema multiforme y livedo reticularis. Las lesiones cutáneas suelen tener una distribución simétrica afectando a áreas acras y de declive, inicialmente pueden no ser purpúricas y en su progresión desde pequeñas pápulas purpúricas pueden aumentar de tamaño y evolucionar hasta formar placas de varios centímetros y desarrollar vesículas y ulceración.
Etiología: El mecanismo principal de producción es el depósito de inmunocomplejos. Estos inmunocomplejos pueden activar el sistema de complemento, produciendo la fracción C3 y C5, lo que produce la quimiotaxis de polimorfonucleares, los cuales liberan enzimas lisosomales que producen el daño tisular. El origen de los complejos Ag-Ac es idiopática en el 50% de los casos. Un 20% se asocian a infecciones especialmente víricas y bacterianas, un 22% se asocian a medicaciones y un porcentaje menor a enfermedades del tejido conectivo (12%) y a antigenos tumorales (<5 br="" de="" especialmente="" linfoproliferativo="" mieloproliferativo.="" nbsp="" o="" origen="">Histologia: El estudio histológico de las lesiones de púrpura palpable demuestra una vasculitis leucocitoclástica que consiste en la presencia de inflamación centrada en un vaso con edema endotelial, necrosis fibrinoide de los vasos capilares, e infiltrado inflamatorio de predominio polimorfonuclear, con fragmentación de los núcleos (leucocitoclasia), hemorragia y trombosis. Los estudios de inmunofluorescencia directa pueden demostrar la presencia de inmunoglobulinas, complemento y firbrinogeno en los vasos dérmicos.
Subgrupos de vasculitis leucocitoclástica cutánea: Existen varias formas clínicas de vasculitis leucocitoclástica cutánea que se pueden agrupar separadamente y que están resumidas en la tabla 10.
- Síndrome de Henoch-Schonlein: Es la forma más frecuente de vasculitis en niños. Se caracteriza por el desarrollo de rash purpúrico, artralgia/artritis, afectación gastrointestinal y nefritis. Las lesiones clínicas son polimorfas las lesiones más características son de púrpura palpable, pero pueden desarrollar pápulas, urticaria, angioedema, vesículas, necrosis y livedo reticulares. Las lesiones afectan principalmente a miembros inferiores y glúteos. Las lesiones cursan a brotes. Ocasionalmente pueden acompañarse de fiebre, malestar, artralgias o mialgias. En general suele reservarse la clasificación en este síndrome para aquellos casos en que se demuestra la presencia de inmunocomplejos circulantes de clase IgA o la presencia de depósitos inmunes en piel de clase IgA
- Urticaria vasculitis: se caracteriza por el desarollo de lesiones cutáneas en forma de habón que a diferencia de lo que ocurre en la urticaria aguda tienen a perdurar con duración de más de 24 horas, deja pigmentación residual, generalmente se asocia con sensación de quemazon más que con picor, suele asociar síntomas sistémicos como fiebre, angioedema, artralgias y artritis y dolor abdomina. Hasta un 30% de pacientes asocian hipocomplementemia y su presencia se asocia con una mayor gravedad.
- Vasculitis crioglobulinemica: Las crioglobulinas son inmunoglobulinas que precipitan con el frio y que producen daño orgánico por dos mecanismos principales, la oclusión vascular (síndrome de hiperviscosidad, principalmente en la crioglobulinemia tipo I y por mecanismos inmunes principalmente en las crioglobulinemias mixtas. Existen 3 tipos básicos de crioglobulinemias, el tipo I monoclonal IgM o IgG, el tipo II mixta monoclonal IgM y polilclonal IgG y el tipo III mixta policlonal IgG e IgM. Las crioglobulinas se relacionan con diversas patologías que incluyen infecciones, enfermedades autoinmunes y neoplasias siendo la más frecuente la infección por virus de la hepatitis C. El porcentaje de pacientes con crioglobulinemia que desarrolla lesiones clínicas varía desde el 2 al 50% de los pacientes, siendo los hallazgos clínicos más característicos la triada de púrpura, artralgias y debilidad que son las manifestaciones debutantes en el 80% de los pacientes, siendo la púrpura la manifestación más característica. Otras manifestaciones cutáneas incluyen úlceras, generalmenet alrededor del maleolo y lesiones isquémicas. El desarrollo de úlceras cutáneas y lesiones de gangrena digital empeoran el pronóstico con un mayor riesgo de infección, sepsis y muerte.
5.2. Vasculitis sistémica primaria asociadas a ANCA
Aproximadamente un 5% de pacientes que se presentan con vasculitis cutánea tienen una vasculitis sistémica y alrededor de un 3% tienen una vasculitis sistémica asociada a ANCA. Estas formas de vasculitis consitutuyen un grupo de enfermedades afectando a vasos de pequeño y mediano calibre que incluyen la granulomatosis de Wegener, la poliangeitis microscópica y el Síndrome de Churg-Strauss. Los ANCA son anticuerpos dirigidos contra antígenos de los polimorfonucleares. Existen 2 patrones de ANCAS : el patrón citoplasmático que incluye los anticuerpos contra la proteinasa 3, y el patron periférico dirigido contra la mieloperoxidasa. En la actualidad se piensa que ciertas moléculas proinflamatorias como el TNF-α y la IL-1 inducen la translocación de la proteinasa 3 y la mieloperoxidasa hacia la superficie de los neutrófilos. Estos antígenos se unen a los ANCA, activando a los neutrofilos y aumentando su adherencia a las células endoteliales dando lugar al daño vascular.
- granular citoplásmico (C-ANCA, PR3-ANCA, con especificidad ante el antígeno citoplásmico proteinasa 3)
- patrón perinuclear (p-ANCA, MPO-ANCA, con especificidad contra el antígeno mieloperoxidasa)
5.2.1.Granulomatosis de Wegener (GW): La granulomatosis de Wegener es una vasculitis sistémica primaria caracterizada por la triada clínica de vasculitis granulomatosa del tracto superior e inferior, glomerulonefritis y grados variables de vasculitis de pequeño vaso. Los pacientes con GW generalmente presentan síntomas de vías respiratorias superiores, incluyendo sinusitis, obstrucción y perforación nasal que puede dar lugar a una deformidad en silla de montar. Otros síntomas frecuentes incluyen otitis media, dolor de oído y disminución de la capacidad auditiva. La afectación pulmonar puede manifestarse en forma de tos productiva y hemoptisis o puede ser asintomática. La afectación renal en forma de glomerulonefritis determina el pronóstico de los enfermos. Al menos el 50% de los pacientes tienen afectación mucocutánea que puede ser la forma de presentación hasta en un 12% de casos. Las manifestaciones cutáneas pueden ser de 3 tipos: 1)púrpura palpable como manifestación de una vasculitis leucocitoclástica de pequeño vaso, 2) nódulos subcutáneos y úlceras como manifestación de una vasculitis de mediano vaso y 3) lesiones polimorfas que incluyen pápulas y nódulos necróticos en las áreas periarticulares, úlceras a tipo pioderma gangrenoso y lesiones de hiperplasia gingival granulomatosa. La afectación cutánea en la granulomatosis de Wegener se asocia con afectación sistémica activa y progresiva. Los criterios para el diagnóstico de la GW están resumidos en la tabla 8. La granulomatosis de Wegener se asocia de forma específica con la presencia de ANCA con especificidad contra la proteinasa 3, con patrón citoplásmico. en los pacientes con enfermedad activa tiene una alta sensibilidad y especifidad.
5.2.2.Poliangeitis microscópica: Forma de vasculitis de pequeño vaso que se asocia frecuentemente a enfermedad renal rápidamente progresiva (glomerulonefritis necrotizante segmentaria focal), afectación cutánea (>75% púrpura palpable), presencia de anticuerpos contra p-ANCA, en ausencia de granulomas extravasculares y depósitos de inmunocomplejos en la inmunofluorescencia directa. La poliangeitis microscopica tiene un pronostico peor que las otras formas de vasculitis asociadas a ANCAS con una supervivencia a los 5 años del 45%. .
5.2.3.Vasculitis granulomatosa y alérgica de Churg-Strauss: La enfermedad de Churg-Strauss es una vasculitis sistémica primaria rara que se observa en pacientes con asma. Se caracteriza por afectar a vasos de tamaños variables con formación de granulomas intra y extravasculares -con intensa presencia de eosinófilos en el infiltrado- y por afectar a pacientes con historia de asma, atopia y eosinofilia periférica. El síndrome de Churg-strauss está considerado resultado de una reacción de hipersensibilidad tipo I, en el cual la proliferación de linfocitos CD4+, TH2 estimulada por diversos alergenos -inhalados, vacunas, medicaciones o infecciones. Los linfocitos TH2 producen interleucina 4, 5 y 13 que estimulan la acumulación de mastocitos, basófilos y especialmente eosinófilos que producen el daño tisular.
Suele asociar sintomatología sistémica con fiebre, mal estado general y pérdida de peso. La manifestación clínica principal e inicial es el desarrollo de asma, que suele afectar a personas con antecedentes de atopia (bronquitis asmática, rinitis o conjuntivitis alérgica y dermatitis). En un segundo estadio las crisis asmáticas se agravan y suelen acompañarse de infiltrados pulmonares con alteraciones radiológicas y eosinofilia periférica y en un tercer estadio se desarrollan signos y síntomas propios de una vasculitis, solo en este último estadio es posible realizar el diagnóstico del síndrome. El 50% de los pacientes fallecen por afectación cardíaca de la enfermedad. El 70% de los pacientes desarrollanlesiones cutáneas en forma de nódulos cutáneos y subcutáneos, denominados granulomas extravasculares de Churg-Strauss, que pueden observarse tanto en la vasculitis de Churg Strauss como en otras enfermedades reumáticas. Otras lesiones cutáneas incluyen el desarrollo de púrpura palpable y livedo reticularis. El 75% de los pacientes desarrollan manifestaciones neurológicas. La analítica más característica muestra una eosinofília periférica con valores de entre 5 y 10x109 eosinófilos/L. El nivel de eosinofilia es un buen marcador para el tratamiento de la enfermedad. El diagnostico de vasculitis puede sugerirse ante la presencia de asma, eosinofilia, infiltrados pulmonares y datos de afectación multisistémica en pacientes con antecedentes atópicos. Los criterios diagnósticos del Síndrome de Churg-Strauss están resumidos en la tabla 9.(ver caso NEJM )
5.3.Arteritis temporal (Vasculitis de células gigantes): La arteritis temporal es la forma de vasculitis sistémica primaria de causa desconocida que afecta a arterias de calibre medio y gran calibre afectando especialmente a las ramas extracraneales de la carótida. Afecta especialmente a personas mayores de 50 años. Cursa con afectación segmentaria de los vasos, con presencia de un infiltrado mononuclear -linfocítico y histiocítico- afectando especialmente a la capa media de las arterias. Se clasifica dentro de las vasculitis mediadas por linfocitos T, observándose en los vasos un infiltrado de predomino CD4, Th1. Clínicamente se caracteriza por síntomas asociados al proceso inflamatorio sistémico (fiebre, malestar general, anorexia, depresion) y manifestaciones clínicas que reflejan la isquemia tisular debida a la estenosis del vaso (cefalea, ceguera, tumefacción y ulceración del cuero cabelludo, claudicación mandibular). Más de 2/3 de los pacientes tienen cefalea que es también el sintoma inicial más frecuente, esta cefalea suele ser de inicio brusco, intensa y de predominio temporal. El examen físico puede mostrar una arteria temporal que se puede visualizar con facilidad en forma de un cordón, doloroso y nodular con el pulso disminuido o ausente . Los criterios diagnósticos están resumidos en la tabla 6. Un 50% de los pacientes asocia polimialgia reumática, síndrome estrechamente relacionado con la arteritis temporal y caracterizado por mialgias, tumefacción, dolor articular, fiebre y pérdida de peso. Las lesiones cutáneas son raras y consisten en el desarrollo de úlceras necróticas en el territorio de la temporal y se asocian a una marcada estenosis de la luz vascular. El diagnóstico debe realizarse en base a los hallazgos clínicos y confirmarse mediante la biopsia de la arteria temporal. La imagen histológica clásica se observa en el 50% de los pacientes y muestra un infiltrado granulomatoso de las capas media e intima de la arteria temporal. Otro 50% de pacientes no muestra los hallazgos característicos, observándose una panarteritis con un infiltrado inflamatorio mixto, con presencia de células mononucleadas y escasos neutrófilos y eosinófilos. Una vez sospechado el diagnóstico debe iniciarse el tratamiento de forma precoz para evitar las complicaciones oculares (la pérdida completa o parcial de la visión de uno o ambos ojos afecta al 20% de los pacientes y es un síntoma presente en las fases iniciales de la enfermedad, -un 44% presentan episodios de amaurosis fugax). La buena y rápida respuesta al tratamiento se considera también criterio diagnóstico de esta entidad.
5.4.Poliarteritis nodosa (PAN): La poliarteritis nodosa es una vasculitis sistémica primaria en la que existe una vasculitis necrotizante de arterias de pequeño y mediano calibre, con marcada afectación renal y visceral. La afectación es segmentaria y tiene predilección por las zonas de bifurcación vascular. Afecta principalmente a varones de mediana edad. Con frecuencia asocia manifestaciones sistémicas generales como fiebre, mal estado general y pérdida de peso. El 70% tienen afectación renal (en forma de arteritis y/o glomerulonefritis, siendo esta la principal causa de muerte), el 60% tienen afectación neurológica periférica con un patrón de afectación de mononeuritis múltiple o polineuropatía. El 50% tienen afectación cutánea incluyendo el desarrollo de nódulos, úlceras y livedo reticularis. En la PAN no existe afectación de las arterias pulmonares. El origen de la PAN es desconocido, se considera dentro de las vasculitis mediadas por inmunocomplejos (reacción tipo III). Se han detectado antígenos de la hepatitis B hasta en un 50% de los pacientes. El hallazgo de inmunocomplejos depositados a nivel de la pared vascular diferencia la PAN de la poliangeitis microscópica. Los cambios histológicos consisten en una inflamación transmural pleomorfica con polimorfonucleares, necrosis fibrinoide, hemorragia y formación de aneurismas. Los criterios diagnósticos de la PAN están resumidos en la tabla 7.
Existe una forma de poliarteritis nodosa limitada a la piel, poliarteritis nodosa cutánea , de curso benigno, donde la afectación visceral está ausente, siendo las manifestaciones más frecuentes el desarrollo de nódulos cutáneos, livedo reticularis y ulceración cutánea.
Existe una forma de poliarteritis nodosa limitada a la piel, poliarteritis nodosa cutánea , de curso benigno, donde la afectación visceral está ausente, siendo las manifestaciones más frecuentes el desarrollo de nódulos cutáneos, livedo reticularis y ulceración cutánea.
6.Actitud diagnóstica ante una vasculitis con manifestaciones cutáneas
Con frecuencia los pacientes con vasculitis debutan con lesiones cutáneas, especialmente púrpura palpable. Las manifestaciones clínicas y patológicas de las vasculitis en piel no son específicas para ningún tipo de vasculitis, por ejemplo, la púrpura palpable que es resultado de la inflamación de un vaso en dermis puede ser originada por una vasculitis infecciosa (pe vasculitis por neiseria), por una vasculitis mediada por inmunoclomplejos ( vgr: la enfermedad del suero, las vasculitis por crioglobulinemia, o la púrpura de Henoch Schonlein), por una vasculitis asociada a ANCA ( granulomatosis de Wegener, poliangeritis microscópica,), una vasculitis alérgica (por una reacción medicamentosa) una vasculitis asociada a un enfermedad reumática (vgr: lupus eritematoso, arteritis reumatoide y síndrome de Sjogren) . De la misma manera, los nódulos cutáneos y subcutáneos inflamatorios pueden ser originados por una variedad de vasculitis, incluyendo la poliarteritis nodosa, la poliangitis microscópica, la granulomatosis de Wegener, el síndrome de Churg-strauss. Cuando existe la sospecha clínica de que estamos ante una vasculitis los pacientes deben ser estudiados de forma minuciosa para determinar la etiología de la vasculitis, la extensión de la afectación vascular y para establecer el tratamiento adecuado.Los estudios clínicos a realizar cuando se sospecha el diagnóstico de vasculitis cutánea deben estar dirigidos de la siguiente manera (Figura 1). Se han descrito diversos algoritmos diagnósticos que deben ser aplicados en grupos grandes de pacientes para comprobar su capacidad diagnóstica.
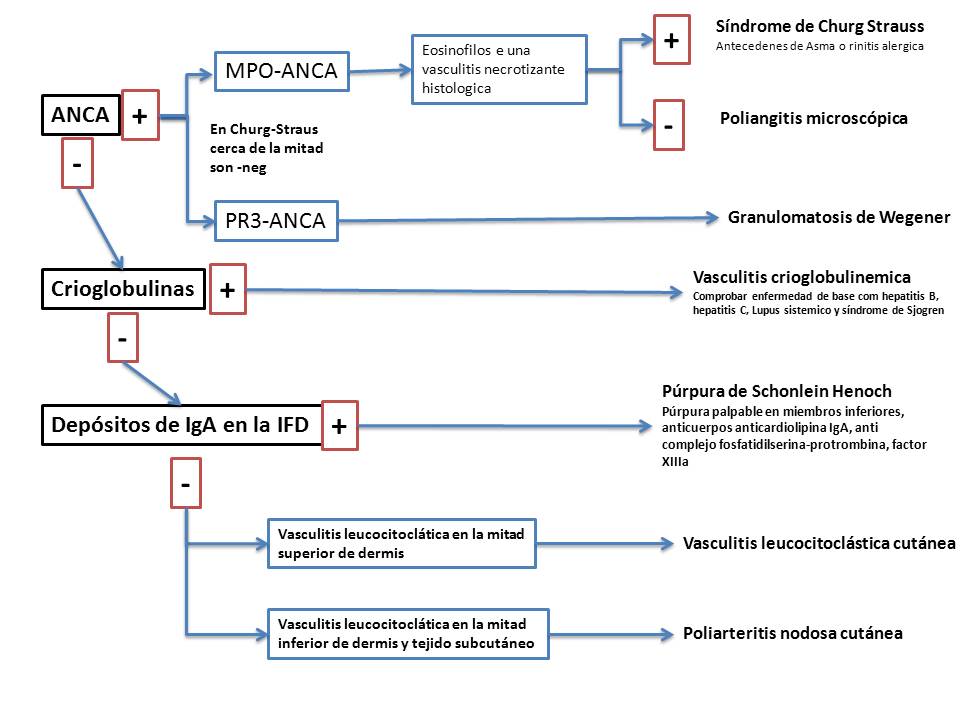{kind=link}
- Confirmar el diagnóstico de vasculitis mediante la realización de una biopsia cutánea para tener el diagnóstico definitivo de vasculitis, ver el tipo de infiltrado, el tamaño del vaso afecto y realizar estudios de inmunofluorescencia directa para demostrar la presencia de inmunocomplejos.
- Examinar la afectación de los órganos sistémicos para establecer si existe afectación cutánea o sistémica.
- Estudiar las posibles causas, intentado determinar si es una vasculitis primaria (dentro de los síndromes de vasculitis primarios) o secundaria y establecer el tratamiento.
Pasos a realizar ante una vasculitis cutánea

- Descartar una causa infecciosa : dado que el tratamiento de las vasculitis por infección directa de los vasos (vasculitis séptica) es completamente diferente de las vasculitis mediada inmunológicamente, esta causa debe ser descartada en el inicio de la valoración de un enfermo con vasculitis.
- Si se determina que es una vasculitis mediada inmunológicamente debe determinarse si es de tipo III, mediada por inmunocomplejos y deben buscarse el origen de estos inmunocomplejos, que puede ser exógeno (medicación, infección, proteínas) o exógeno (DNA, inmunoglobulinas, antígenos tumorales). La determinación de la existencia de la formación de inmunocomplejos como causa de la vasculitis es tranquilizadora ya que aumenta las posibilidades de que la vasculitis sea autolimitada y facilita la retirada de la fuente del antígeno (bien por retirada de la medicación o por tratamiento de la enfermedad de base).
- En relación a la clínica del paciente, se establecerá una serie de exploraciones para determinar la afectación sistémica. Los signos y síntomas que sugieren vasculitis en otros órganos
- Afectación muscular: Mialgias con elevación de enzimas musculares
- Afectación digestiva: Dolor abdominal con sangre oculta en feces o elevación de enzimas pancreáticos
- Afectación cardiaca: Angina con elevación de enzimas micocardicos
- Afectación renal: Hematuria con proteinuria
- Afectación de nervios periféricos: Mononeuritis múltiple con defectos en la conducción nerviosa
- Afectación de sistema nervioso central: Disfunción cerebral o visual
- Test serológicos: ANA, Crioglobulinas, Anticuerpos anti hepatitis B y C, ANCA, y Niveles de complemento.
Tratamiento de las vasculitis leucocitoclástica cutánea
En muchos pacientes la vasculitis de pequeño vaso va a tener un curso relativamente benigno, autolimitado, especialmente si la enfermedad está limitada a la piel. Sin embargo, los pacientes con enfermedad agresiva como los pacientes con vasculitis de pequeño vaso asociado a ANCAS necesita iniciar el tratamiento de forma rápida y agresiva.1-De soporte: reposo, elevación de las partes declives y protección frente a traumatismos y frío
2-Antiinflamatorios: corticoides tópicos, antiinflamatorios no esteroideos: Indometacina,
3-Antiagregantes plaquetarios: aspirina, dipiridamol.
4-Sistémicos: corticoides sistémicos y medicaciones inmunosupresoras.
casos per a discussió a classe
bibliografia
- ANCA associated vasculitis
- crioglobulinemias
- Gonzalez-Gay, MA, Garcia-Porrua C, Pujol RM. Clinical approach to cutaneous vasculitis. Curr Op Rheumatol, 2005, 17:56
- Carlson JA. The histological assessment of cutaneous vasculitis. Histopathology 2010 56:3-23
- Carlson JA; Chen KR. Cutaneous vasculitis update: small vessel neutrophilic vasculitis syndromes. Am J Dermatopath 2006, 28:486
- Kawakami T. New algorithm (kawakami algorithm) to diagnose primary cutaneous vasculitis. J dermatolo. 2010 37:113-124
- Bosch X; Guilabert A; Font J. Antineutrophil cytoplasmic antibodies. Lancet 2006; 368(9533):404-418.
- Jennette JC, Falk RJ. Small vessel vasculitis. N Eng J Med 1997, 337:1512
- Ledford DK. Immunologic aspects of vasculitis and cardiovascular diseases. JAMA 1997, 278:1962.
- Lotti T, y col. Cutaneous small-vessel vasculitis. J Am Acad Dermatol 1998, 39:667.
- Buerguer disease
- petechial rash on the extremities. cutaneous leucocitoclastic vasculitis
- Kawasaki disease