sábado, 25 de mayo de 2013
L- GLUTAMINA.
De Whikipedia.
Este aminoácido en suplemento dietético sírve para que los músculos ejercitados no bajen de volumen. En ciertas ocasiones, como el estrés, traumas o infecciones, puede ser considerado como "semiesencial". Se emplea en casos en los que algunas dolencias han postrado a un paciente en cama durante un periodo largo de tiempo, los atletas de musculación lo emplean debido a sus efectos "constructores" de músculo, así como en los pacientes de cancer y sida.2 Esto es debido a que las situaciones de trauma, cirugía y demás situaciones de estrés hacen que los músculos liberen glutamina al torrente sanguíneo con la consiguiente pérdida de masa muscular.
La suplementación de L-glutamina puede ser beneficiosa en casos de artritis, enfermedades inmunodeficientes, fibrosis, desordenes intestinales, úlceras pépticas, daños en los tejidos debido a radiación, cancer (en algunos casos se detectan niveles anormales submínimos de glutamina) , etc. La glutamina se comercializa en polvo y en capsulas. Los suplementos deben guardarse en un ambiente seco ya que la humedad favorece la hidrólisis de la glutamina en amoníaco y en ácido piroglutámico.
La glutamina no debería ser administrada a personas con cirrosis, problemas renales, síndrome de Reye,6 o cualquier otro problema de salud resultante de un exceso de amoniaco en sangre. Investigaciones realizadas en animales han mostrado que la administración de glutamina tiene efectos de reducción del apetito, sin embargo este efecto no se ha estudiado en humanos.7 Se ha administrado con éxito en la nutrición parenteral de los pacientes hospitalarios (en dosis estándares, que van desde los 2.8 hasta los 7.3 gramos/1000 calorías).
L-Glutamina es biosintetizada por el hígado y los pulmones. Durante el ejercicio de musculación intenso se liberan ciertas cantidades de glutamina superiores a las cantidades que sintetiza el cuerpo humano. Los estudios científicos han demostrado que este consumo desgasta las reservas naturales de glutamina en los músculos,8 es por esta razón por la que ciertos atletas lo emplean como suplemento dietético. Por ejemplo los atletas que realizan ejercicio anaeróbico liberan cerca de un 45% comparado con los niveles anteriores a la realización del ejercicio. Cuando los mismos atletas prosiguen con ejercicio aeróbico durante 10 días, su concentración de glutamina en el plasma desciende hasta un 50%. El descenso en los niveles de glutamina se mantiene incluso seis días tras la recuperación del ejercicio. Estos datos sugieren que este tipo de atletas necesita una suplementación de glutamina en sus dietas capaz de reponer las reservas en los músculos. Los atletas que someten a un sobre-esfuerzo a sus músculos (sin un adecuado reemplazo de glutamina) incrementan su riesgo de infección y a menudo se recuperan más lentamente de los daños sufridos.8
martes, 14 de mayo de 2013
TRATAMIENTO DEL TDAH
Los niños que tienen TDAH pueden mejorar
con tratamiento, pero no hay cura. Los síntomas
del TDAH expresan un problema biológico y
por lo tanto se abordan más eficazmente con
tratamiento farmacológico, el objetivo seria hacer desaparecer los síntomas a largo plazo para
posibilitar el desarrollo social, intelectual y afectivo de ese niño y ayudarle a desarrollar técnicas
que contrarresten sus limitaciones.
Los tratamientos habituales se basan paradójicamente en estimulantes, que tienen un efecto
tranquilizante en personas con este trastorno.
Actualmente, las sustancias más empleadas en
Estados Unidos son el metilfenidato (Ritalina®)
y la d-L anfetamina (Adderall®), seguidas de la
dexanfetamina y la metanfetamina. Recientemente se aprobó el uso de la prodroga lisdexanfetamina (Vyvanse®) que al ser una amalgama
de la dexanfetamina con el aminoácido lisina,
consigue prolongar su acción terapéutica, demorando la metabolización de la sustancia.
En España el único estimulante aceptado para el
tratamiento del TDAH es el metilfenidato, comercializado con el nombre de Rubifen® (efecto
inmediato) o Concerta® (liberación prolongada).
Otro fármaco disponible es la atomoxetina
(Strattera®), inhibidor de la recaptación sináptica
de la norepinefrina, al tratarse de un principio
activo adrenérgico no estimulante, se postula
como fármaco de segunda línea cuando los estimulantes no son bien tolerados.
Aunque los estimulantes son la primera línea en
la terapéutica de este trastorno, algunos agentes
antidepresivos han mostrado cierta utilidad, sobre todo cuando el TDAH cursa con comorbilidades como el trastorno depresivo o trastornos
de ansiedad.
Investigaciones científicas asocian todos estos
tratamientos con una serie de riesgos y complicaciones. Existen numerosos efectos secundarios que van desde problemas de sueño y dolores de estómago, a toxicidad hepática y fenómenos de despersonalización. Por esta razón, el
tratamiento farmacológico ha sido abiertamente
rechazado por profesionales partidarios de
tratamientos menos agresivos.
Naturalmente, hablar con el médico acerca de
cuál es el mejor tratamiento para el niño sería
el paso inicial, pero a su vez se recomienda un
tratamiento psicológico a través de una terapia
conductual para reducir las conductas disruptivas del niño en los diferentes ambientes, ense-
ñándolos así a controlar su comportamiento
para que puedan desempeñarse mejor en la
escuela y en casa. Igualmente puede ser aconsejable una intervención psicopedagógica sobre
los problemas de aprendizaje que suelen aparecer en gran parte de los sujetos con TDAH.
Actualmente se están desarrollando terapias de
desarrollo positivo en los niños, que intentan
reforzar los aspectos potenciales de los jóvenes
mediante deporte y dinámicas de grupo.
Un especialista puede ayudar a los padres a
brindar orientación y comprensión al niño en
el ámbito familiar, al apoyar a su hijo, se ayuda
a todos los miembros de la familia. Se deberían
limitar las distracciones en el ambiente del niño,
verificar que duerma bien y que consuma una
dieta saludable y variada, con mucha fibra y
nutrientes básicos. A su vez, se debería hablar
con la escuela y profesores ya que algunos niños
con TDAH pueden recibir servicios educativos
especiales.
El 2-dimetilaminoetanol, conocido como Deanol
o Deaner®, es un precursor natural del neurotransmisor acetilcolina. Los estudios han demostrado que la colina y los niveles de acetilcolina aumentan con la suplementación de DMAE.
Ya en 1957, Pfeiffer et al., informaron de que la
2-dimetilaminoetanol (DMAE) tenia efectos estimuladores y postulaban la posibilidad de que
fuera un precursor de la acetilcolina cerebral.
Las primeras investigaciones informaban de
que el compuesto era útil en la gestión de
desórdenes de comportamiento de aprendizaje durante la infancia, mejorando la efectividad
académica de los niños con TDAH, otorgando
con estos hallazgos el crédito apropiado a los
que primero describieron los efectos farmacológicos del DMAE, así como los que hacían las
primeras observaciones clínicas de la acción de
este nutriente.
En los años 70 se llevaron a cabo estudios para
determinar su eficacia en el tratamiento de las
alteraciones en el aprendizaje de los niños. Entre 1973 y 1974, Lewis y Young informaron de
un estudio a doble ciego en niños con problemas de TDAH. El estudio randomizado contó
con 74 niños de entre 6 y 12 años asignados a
placebo o DMAE (500mg) durante 10 semanas. Los exámenes neurológicos, psicológicos y
psicométricos fueron llevados a cabo, antes y
después del ensayo. Estos tests encontraron una
mejora significativa en los pacientes que habían
estado tomando DMAE, respecto a los que
habían tomado placebo como a los que habían
tomado metilfenidato.
Desde entonces, no sólo se ha probado la efectividad del DMAE para estos problemas, sino
también, para otras formas de alteraciones
mentales, que incluyen: depresión, falta de coordinación motora y fatiga mental.
Complementos
naturales en el TDHA
DMAE (2-dimetilaminoetanol)
En estudios más recientes como el de Knusel et
al, en 1990, se ha demostrado la evidencia bioquímica de la efectividad del DMAE, en la mejora de las funciones cognitivas, en los pacientes
con la enfermedad de Alzheimer y demencia
degenerativa progresiva.
Los fosfolípidos son importantes componentes bioquímicos de las células del cerebro, así
como de todas las membranas celulares del
organismo.
Los fosfolípidos son lípidos iónicos polares compuestos de 1,2-diacilglicerol y un enlace fosfodiéster que une el esqueleto del glicerol a alguna base, generalmente nitrogenada, tal como
la colina, serina o etanolamina. Los fosfolípidos
más abundantes en los tejidos humanos son
la fosfatidilcolina, la fosfatidiletanolamina y la
fosfatidilserina.
Aunque se hallan presentes en fluidos corporales tales como el plasma y la bilis, los fosfolípidos se encuentran en concentraciones más
elevadas en las diversas membranas celulares
donde realizan muchas funciones diferentes.
La función principal de los fosfolípidos es servir
como componentes estructurales de las membranas de la superficie celular y de los orgánulos
subcelulares. El carácter anfipático de los fosfolípidos les permite su autoasociación a través
de interacciones hidrofóbicas, dejando la parte
hidrofílica hacia el exterior.
Las membranas celulares no son simplemente
paredes que separan el interior del exterior
de la célula, especialmente en el cerebro. El
fenómeno del pensamiento reside, en gran
parte, en la transmisión del impulso eléctrico a
través de las neuronas. Esto requiere la correcta
formación de la bicapa lipídica que envuelve a
las células neuronales, de ahí que la fosfatidilserina, la fosfatidiletanolamina y la fosfatidilcolina,
sean esenciales para la adecuada transmisión de
las señales neuronales. Además, la fosfatidilcolina
y fosfatidilserina son importantes para la óptima
función del cerebro, debido a la variedad de papeles fisiológicos que realizan, incluyendo la producción de neurotransmisores y la producción
de receptores neuronales.
Existe la teoría de que la fosfatidilserina posee
la capacidad de potenciar la función cerebral.
Las membranas celulares son más complejas
que simples barreras alrededor del citosol. Son
estructuras dinámicas, constantemente en movimiento. Incluso las células que parecen estar
estancadas o inmóviles están reemplazando sus
membranas constantemente. Enviadas desde una
estructura que se llama aparato de Golgi, unas
pequeñas burbujas, llamadas vesículas, reemplazan continuamente la membrana celular. Estas vesículas están compuestas de material de la
membrana celular (o fosfolípidos), para ser expulsadas desde el interior. De la misma manera,
a lo largo de la superficie interior de la membrana celular se forman vesículas que contienen nutrientes y materiales necesarios que se encuentran cerca del fluido exterior, para ser llevados
al interior de la célula. De estos procesos de
exocitosis y endocitosis se encarga una sección
especializada del cerebro, permitiendo este proceso dinámico de mantenimiento y la imprescindible comunicación célula a célula, gracias a la
transmisión de señales electroquímicas.
Unas vesículas muy pequeñas, llamadas gránulos,
se llenan con neurotrasmisores, como la acetilcolina o la serotonina. Estas vesículas se acumulan en la superficie interior de las membranas
presinápticas. Cuando una señal eléctrica debe
ser transmitida de una célula nerviosa a otra, la
delgada vesícula se fusiona rápidamente con la
membrana celular presináptica, liberando sus
neurotransmisores a la sinapsis, donde células
nerviosas postsinápticas, a través de receptores,
detectan la presencia de los neurotransmisores
generando una carga eléctrica que se transporta a lo largo de toda su longitud.
La capacidad de la célula nerviosa para desenvolverse en la compleja función de la comunicación,
se basa esencialmente, en las propiedades de las
membranas de la sinapsis. Si la célula no puede
fusionarse con una vesícula llena de neurotransmisores, la señal será lenta. Por otra parte, si
se fusionara demasiado rápido, se podrían producir señales nerviosas cuando no deberían. Los
investigadores creen, que la incorporación de
la fosfatidilserina, a estas membranas cruciales,
puede estabilizar las características fluidas de
la membrana. Cuando hay suficiente fosfatidilserina en la dieta, la liberación de gránulos de
neurotransmisores se produce en el momento
apropiado y a la velocidad adecuada.
Las células cerebrales contienen gran cantidad de fosfolípidos en sus membranas pero su
concentración disminuye con la edad. Si consideramos que la membrana celular posee importantísimas funciones que van desde regular
las sustancias que entran y salen de la célula, la
composición interna de éstas y la transmisión de
señales, podemos asegurar que los fosfolípidos
como la fosfatidilcolina, fosfatidiletanolamina y
en especial la fosfatidilserina son de gran relevancia para la actividad cerebral.
Respecto a los fosfoinosítidos o fosfatidilinositoles, son un tipo de fosfolípidos que contienen
en su estructura uno o más inositoles. No contienen nitrógeno y se enecuentran principalmente en el cerebro. Poseen especial relevancia
en biología celular puesto que, cuando se separa
el fosfolípido del inositol, estos actúan como
segundos mensajeros en la transducción de se-
ñales de célula a célula.
La fosfatidilserina junto con la fosfatidilcolina,
fosfatidiletanolamina y los fosfoinosítidos son
nutrientes que desempeñan un papel vital como
componentes estructurales de las membranas
celulares. Mantiene la integridad estructural y
la fluidez de las membranas y esto se observa
en una mejora del aprendizaje y de la concentración, en una potenciación de la memoria y en
un equilibrio del estado anímico.
Los ácidos omega 3 y omega 6 son ampliamente
comercializados como tratamientos efectivos
para el TDAH. Sin embargo, la base de evidencia para este uso, en el mejor de los casos, no
es concluyente.
El aceite de onagra fracasó en demostrar eficacia en el trastorno del déficit de atención en un
ensayo doble ciego, controlado con placebo. En
otro pequeño ensayo comparativo, controlado
con placebo, el aceite de onagra comprobó ser
menos efectivo que la D-anfetamina.
Respecto a los ácidos grasos omega 3 juegan
un importante papel en el crecimiento y desarrollo de los niños. El ácido docosahexaenoico
o DHA es el ácido graso más insaturado que
existe en la naturaleza siendo un componente
imprescindible para la formación de fosfolípidos necesarios para el mantenimiento de las
membranas celulares. El DHA ha sido utilizado
como terapia coadyuvante para el tratamiento
de TDAH, demostrando pequeñas mejoras en
estudios realizados.
En ocasiones se ha recomendado para el
tratamiento del TDAH el uso de complementos
nutricionales a base de vitamina B3 (niacina), vitamina B6 o complejos vitamínicos y minerales.
Otros suplementos también recomendados
incluyen combinaciones de aminoácidos (usualmente GABA, glicina, L-glutamina, L-fenilalanina,
L-tirosina, taurina), algas, calcio, inositol, hierro,
magnesio, hierba de San Juan, minerales traza y
zinc. Sin embargo, ninguna evidencia científica
apoya alguno de estos tratamientos para la regulación del TDAH en estos momentos.
sábado, 11 de mayo de 2013
INOSITOL EN ALZHEIMER
Tratamiento de inositol de la enfermedad de Alzheimer: un estudio doble ciego, cruzado y controlado con placebo.
Fuente
Centro de Salud Mental Abarbanel, Bat Yam, Israel.
Abstracto
1. Un ensayo cruzado doble ciego controlado de 6 g de inositol al día frente a la glucosa durante un mes cada uno se llevó a cabo en 11 pacientes con Alzheimer. 2. Las puntuaciones globales CAMCOG mostraron una tendencia para una mayor mejora con inositol que no fue significativa. 3.Lengua y orientación mejoraron significativamente más en inositol que con placebo. No hubo efectos secundarios graves. 4. Las dosis más altas de inositol deben ser estudiados en la enfermedad de Alzheimer durante períodos más largos.
viernes, 10 de mayo de 2013
HIPERPROLACTINEMIA EN LA MUJER
Instituto Nacional de Endocrinología Departamento de Reproducción Humana
La prolactina (PRL) es una de las hormonas anterohipofisarias cuya molécula se compone de 198 residuos aminoacídicos con 6 hemicistinas como característica principal, su secreción es pulsátil y circadiana, los valores más elevados se encuentran en la fase "nom-REM" del sueño y durante la fase luteal del ciclo menstrual de la mujer, así como durante la vida fetal a partir de la semana 30 y hasta la primera semana de recién nacidos.1,2
La secreción inadecuada de PRL puede producir en la mujer alteraciones del ciclo menstrual, trastornos de la ovulación, infertilidad y galactorrea. Así surgieron, según el trastorno clínico relacionado con su aparición, varios síndromes identificados por el nombre de los primeros que los describieron, como son: el síndrome de Chiari-Frommel, cuando la triada aparecía después del parto, sin la presencia de tumor hipofisario; cuando el cuadro clínico se presentaba espontáneamente sin tumor hipofisario demostrable se denominó síndrome de Ahumada-Del Castillo y síndrome de Forbes-Albright al aparecer los síntomas asociados a tumor hipofisario.3En la actualidad se prefiere no utilizar estos términos y agruparlos bajo el nombre genérico de hiperprolactinemia.
En este artículo revisaremos los efectos de la hiperprolactinemia sobre el hipotálamo y el ovario, así como su causa, cuadro clínico y diagnóstico.
En las pacientes adolescentes, los síntomas más frecuentes son amenorrea (primaria o secundaria), cefalea, retraso puberal, galactorrea y alteraciones del campo visual,15-17 y en ellas la causa tumoral aparece con mucha frecuencia, lo cual se trata de explicar por un aumento de los estrógenos en la adolescencia, así como la influencia de opiáceos endógenos, sustancias "TRH-similes", péptido intestinal vasoactivo (VIP), histamina, sustancia P, neurotensina y bombesina, que están involucrados en la génesis de estos adenomas en la edad infanto-juvenil.16
La cefalea es otro síntoma común, las pacientes lo describen como penetrante, de tipo frontal, debajo de los ojos; es más frecuente cuando la causa de la hiperprolactinemia es un prolactinoma, aunque puede ser expresión directa del estado hiperprolactinémico sin que necesariamente exista un tumor hipofisario productor de PRL. Este síntoma suele mejorar después del tratamiento.18,19 Otros síntomas, que en ocasiones refieren las pacientes, son la dispareunia y disminución de la libido o frigidez.2,14 El síndrome de tensión premenstrual puede estar presente, así como trastornos del campo visual, que cuando aparecen hacen sospechar la causa tumoral por extensión supraselar y compresión del quiasma óptico.
La hiperprolactinemia puede acompañarse de una caída importante de los valores de estrógenos en la primera fase del ciclo menstrual y provocar un hipogonadismo que, de mantenerse, puede conducir a una osteoporosis.20,21
En una mujer aparentemente normal cuyo único problema es la infertilidad o simplemente los antecedentes de abortos espontáneos repetidos antes de las 12 sem, como expresión de un defecto de la fase lútea,14,22 la hiperprolactinemia puede ser la causa. Otra de las manifestaciones clínicas que se puede presentar en este trastorno es el hirsutismo, como expresión del hiperandrogenismo que se desarrolla por aumento de la producción de andrógenos adrenales y ováricos.1,5 El síndrome de ovarios poliquísticos puede asociarse con hiperprolactinemia,1 pero el mecanismo por el cual la PRL se eleva no está claro, aunque se plantea que está en relación con el aumento de la producción de estrógenos.14 También puede encontrarse hiperprolactinemia sin nin-guna manifestación clínica, por la presencia en sangre de la forma molecular conocida por big-big PRL, por su alto peso molecular ( > 60 000 D), a lo que se denomina macroprolactinemia.23,24
Se intentó establecer pruebas dinámicas para evaluar la secreción de PRL que permitieran distinguir entre hiperprolactinemia funcional y tumoral, pero después de numerosos esfuerzos no se ha podido trazar una separación absoluta entre ambos estados2 por lo que se desecharon estos procederes.
Los estudios del campo visual y la perimetría están indicados en pacientes con macroadenomas y con extensión supraselar del tumor o cuando se obtienen, al realizar el examen oftalmológico, síntomas y signos de afectación de esta esfera.14,28
En las embarazadas con hiperprolactinemia tumoral deben realizarse estudios evolutivos del campo visual para detectar un crecimiento del tumor.14,29
Así, en mujeres que presentan hiperprolactinemia, con todas las causas conocidas excluidas y estudios anatómicos normales, es planteable una hiperprolactinemia idiopática o funcional por lo que éste es solo un diagnóstico de exclusión.30
En conclusión, la causa más frecuente de hiperprolactinemia en la mujer es el adenoma hipofisario productor de prolactina (prolactinoma), los síntomas de más valor diagnóstico (amenorrea y galactorrea) están relacionados con el trastorno hormonal, aunque en casos de macroprolactinomas pueden presentarse manifestaciones neurooftalmológicas. Además de la determinación de prolactina, los estudios de mayor valor diagnóstico son los imagenológicos pues las pruebas dinámicas no son útiles. El estudio del campo visual es útil en el diagnóstico del macroprolactinoma y en la valoración evolutiva de los pacientes que lo padecen.
Hiperprolactinemia en la mujer: Causas, cuadro clínico y diagnóstico
Dr. Enrique J. Perdomo Estrada,1 Dr. Felipe Santana Pérez2 y Dr. Rubén S. Padrón Durán3- Especialista de I Grado en Endocrinología.
- Maestro en Ciencias en Salud Reproductiva. Especialista de II Grado en Endocrinología. Investigador Auxiliar.
- Doctor en Ciencias Médicas. Especialista de II Grado en Endocrinología. Investigador y Profesor Titular.
RESUMEN
Se hizo una revisión de los trabajos más actualizados sobre la hiperprolactinemia por ser ésta una causa frecuente de amenorrea, galactorrea e infertilidad en las mujeres en edad reproductiva. Los adenomas productores de prolactina son los tumores más frecuentes de la hipófisis y entre éstos predominan los microadenomas. Los síntomas de los prolactinomas están más relacionados con la hiperprolactinemia, pero en casos de macroadenomas pueden presentarse síntomas de compromiso local como cefalea, defectos del campo visual y, más raramente, síntomas de compresión del seno cavernoso. Las pruebas de función dinámica propuestas para el diagnóstico diferencial entre las causas idiopática y tumoral de la hiperprolactinemia no son útiles de manera individual, sólo pudieran usarse para discriminación grupal. Después de confirmado el diagnóstico de hiperprolactinemia deberá buscarse la presencia de una lesión compatible con un tumor hipofisario a través de la tomografía axial computadorizada contrastada o la resonancia magnética nuclear de silla turca. Nuestro objetivo con esta revisión teórica ha sido mostrar las principales causas, el cuadro clínico y los procederes diagnósticos en esta entidad que constituye en la actualidad un importante motivo de consulta en esta especialidad.Descriptores DeCS: HIPERPROLACTINEMIA/etiología; HIPERPROLACTINE-MIA/diagnóstico.La prolactina (PRL) es una de las hormonas anterohipofisarias cuya molécula se compone de 198 residuos aminoacídicos con 6 hemicistinas como característica principal, su secreción es pulsátil y circadiana, los valores más elevados se encuentran en la fase "nom-REM" del sueño y durante la fase luteal del ciclo menstrual de la mujer, así como durante la vida fetal a partir de la semana 30 y hasta la primera semana de recién nacidos.1,2
La secreción inadecuada de PRL puede producir en la mujer alteraciones del ciclo menstrual, trastornos de la ovulación, infertilidad y galactorrea. Así surgieron, según el trastorno clínico relacionado con su aparición, varios síndromes identificados por el nombre de los primeros que los describieron, como son: el síndrome de Chiari-Frommel, cuando la triada aparecía después del parto, sin la presencia de tumor hipofisario; cuando el cuadro clínico se presentaba espontáneamente sin tumor hipofisario demostrable se denominó síndrome de Ahumada-Del Castillo y síndrome de Forbes-Albright al aparecer los síntomas asociados a tumor hipofisario.3En la actualidad se prefiere no utilizar estos términos y agruparlos bajo el nombre genérico de hiperprolactinemia.
En este artículo revisaremos los efectos de la hiperprolactinemia sobre el hipotálamo y el ovario, así como su causa, cuadro clínico y diagnóstico.
EFECTOS DE LA HIPERPROLACTINEMIA SOBRE EL HIPOTALÁMO Y LOS OVARIOS
La hiperprolactinemia ocupa un lugar de importancia entre las causas de trastornos de la ovulación e infertilidad en la mujer; aunque no se conoce con exactitud el mecanismo por el cual se produce, se plantea que los altos niveles de PRL en sangre, podrían provocar anovulación por bloqueo del pulso de la hormona luteinizante (LH) y por interferencia en el efecto del mecanismo de retroalimentación positiva del estradiol (E2) al nivel hipotalámico, mediante el bloqueo de los receptores de estrógenos.1,2 En el ovario, la hiperprolactinemia puede provocar disminución del número o de la afinidad de los receptores de LH en el cuerpo lúteo, lo cual se asocia a una disminución en la producción y secreción de progesterona y podría explicar el hallazgo clínico de mujeres infértiles con deficiencia de la fase luteal y niveles bajos de progesterona.2CAUSA
Existen múltiples situaciones en las cuales los niveles de PRL se elevan de forma transitoria o permanente; cuando esto se produce por causas como: el sueño, el embarazo, la lactancia, la manipulación de la mama, la anestesia, el ejercicio, la cirugía y el coito con orgasmo, estamos en presencia de causas fisiológicas de hiperprolactinemia.4El tumor hipofisario productor de PRL es el más frecuente de los tumores de esta glándula y por tanto, al diagnosticar una hiperprolactinemia constituye una obligación buscar su presencia.4 También debemos investigar otras causas que incrementan las cifras de PRL, como son: el uso de psicofármacos, estrógenos u otros medicamentos, la presencia de hipotiroidismo primario.1,4,5 En este último se produce una hiperplasia de las células hipofisarias como respuesta a la acción de la TRH que no sólo estimula a las células tirotropas sino también a las lactotropas lo cual explicaría, la existencia de hiperprolactinemia.4 Se ha descrito además esta entidad en pacientes con hipotiroidismo subclínico y asociación de hipotiroidismo primario y tumor productor de PRL en un mismo paciente.6 Recientemente se comunicaron casos de hiperprolactinemia con aumento de tamaño de la silla turca, en los cuales hubo exposición prenatal al dietiletilbestrol y se sugirió una posible relación causal.4 La hipofisitis linfocítica4,7 también se ha descrito entre las causas de hiperprolactinemia, por sí sola o asociada a otras endocrinopatías "linfocíticas" como la tiroiditis linfocítica, enfermedad de Hashimoto y paratiroiditis.1,4 Otros han hallado hiperprolactinemia en pacientes con tumor dermoide del ovario,8 en el curso de leucemia mieloide aguda,9 en la esclerosis múltiple,10 en la psoriasis11 y en el lupus eritematoso sistémico;12 así como la enfermedad de Cushing, por un tumor mixto productor de ACTH y PRL.13MANIFESTACIONES CLÍNICAS
La amenorrea y la galactorrea son las manifestaciones más importantes para sospechar una hiperprolactinemia desde el punto de vista clínico,14 pueden aparecer después de un embarazo o sin relación con éste. La amenorrea secundaria es el trastorno menstrual más llamativo para el diagnóstico clínico, pero la oligomenorrea, la amenorrea primaria y la hiperpolimenorrea son otras manifestaciones de la esfera ginecológica que se presentan con frecuencia.2,14La galactorrea puede ser unilateral o bilateral, espóntanea o presentarse sólo por la manipulación de la mama, pero su ausencia no niega la hiperprolactinemia ni su presencia la afirma.14 Cuando la galactorrea aparece asociada a amenorrea secundaria o primaria, el diagnóstico de hiperprolactinemia es muy probable y la posibilidad de un tumor hipofisario es mayor que si aparecieran estos síntomas aislados.14-17En las pacientes adolescentes, los síntomas más frecuentes son amenorrea (primaria o secundaria), cefalea, retraso puberal, galactorrea y alteraciones del campo visual,15-17 y en ellas la causa tumoral aparece con mucha frecuencia, lo cual se trata de explicar por un aumento de los estrógenos en la adolescencia, así como la influencia de opiáceos endógenos, sustancias "TRH-similes", péptido intestinal vasoactivo (VIP), histamina, sustancia P, neurotensina y bombesina, que están involucrados en la génesis de estos adenomas en la edad infanto-juvenil.16
La cefalea es otro síntoma común, las pacientes lo describen como penetrante, de tipo frontal, debajo de los ojos; es más frecuente cuando la causa de la hiperprolactinemia es un prolactinoma, aunque puede ser expresión directa del estado hiperprolactinémico sin que necesariamente exista un tumor hipofisario productor de PRL. Este síntoma suele mejorar después del tratamiento.18,19 Otros síntomas, que en ocasiones refieren las pacientes, son la dispareunia y disminución de la libido o frigidez.2,14 El síndrome de tensión premenstrual puede estar presente, así como trastornos del campo visual, que cuando aparecen hacen sospechar la causa tumoral por extensión supraselar y compresión del quiasma óptico.
La hiperprolactinemia puede acompañarse de una caída importante de los valores de estrógenos en la primera fase del ciclo menstrual y provocar un hipogonadismo que, de mantenerse, puede conducir a una osteoporosis.20,21
En una mujer aparentemente normal cuyo único problema es la infertilidad o simplemente los antecedentes de abortos espontáneos repetidos antes de las 12 sem, como expresión de un defecto de la fase lútea,14,22 la hiperprolactinemia puede ser la causa. Otra de las manifestaciones clínicas que se puede presentar en este trastorno es el hirsutismo, como expresión del hiperandrogenismo que se desarrolla por aumento de la producción de andrógenos adrenales y ováricos.1,5 El síndrome de ovarios poliquísticos puede asociarse con hiperprolactinemia,1 pero el mecanismo por el cual la PRL se eleva no está claro, aunque se plantea que está en relación con el aumento de la producción de estrógenos.14 También puede encontrarse hiperprolactinemia sin nin-guna manifestación clínica, por la presencia en sangre de la forma molecular conocida por big-big PRL, por su alto peso molecular ( > 60 000 D), a lo que se denomina macroprolactinemia.23,24
DIAGNÓSTICO
En toda paciente con galactorrea, trastornos menstruales, hirsutismo, disminución de la libido o infertilidad, deben determinarse los niveles de PRL plasmática, entre el tercer y quinto día del ciclo menstrual, si este es regular o en cualquier momento, si presenta amenorrea u oligomenorrea.2 Si en la primera determinación se obtienen cifras elevadas debe repetirse y si se confirma nuevamente, se establece el diagnóstico de hiperprolactinemia. Una historia clínica com-pleta, con un interrogatorio y examen físico detallados, permitirá orientarnos hacia la causa de la hiperprolactinemia. La primera causa que se debe descartar antes de emprender otros estudios, es el embarazo. El uso de estrógenos, anticonceptivos orales y drogas que aumentan la secreción de PRL debe precisarse en el interrogatorio, así como la presencia de quemaduras u otras lesiones en tórax que se buscan en el examen físico. Una vez comprobado lo anterior se investigará la existencia de un hipotiroidismo primario.2,14Cuando no existe un hipotiroidismo primario, es obligada la búsqueda de un tumor hipofisario. Los estudios imagenológicos simples de la silla turca fueron los primeros en utilizarse; en particular, las vistas lateral y frontal de cráneo y selectiva de la silla turca. La politomografía de la silla turca, es otro de los estudios imagenológicos utilizados para el diagnóstico de tumores hipofisarios. En estos estudios se pueden hallar deformidades y aumento de tamaño de la silla turca, erosión del piso, destrucción de las clinoides posteriores, así como destrucción total de la silla turca; imágenes que sugieren la presencia de un tumor hipofisario. La tomografía axial computadorizada (TAC) vino a revolucionar el campo de la radiología y el uso de contrastes ha permitido establecer con mayor certeza el diagnóstico de tumores pequeños y, por tanto, muchas de las hiperprolactinemias antes consideradas funcionales o idiopáticas hoy se diagnostican como tumores.25,26 La resonancia magnética nuclear (RMN) es otro de los estudios imagenológicos que se utilizan hoy para detectar la pre-sencia de un tumor hipofisario y algunos investigadores la consideran más específica en el diagnóstico de microadenoma que la TAC.25,27 Los equipos más modernos de TAC y RMN pueden realizar cortes de 1-2 mm, utilizan menos radiación que la politomografía y pueden detectar tanto extensión supraselar como paraselar del tumor.14Se intentó establecer pruebas dinámicas para evaluar la secreción de PRL que permitieran distinguir entre hiperprolactinemia funcional y tumoral, pero después de numerosos esfuerzos no se ha podido trazar una separación absoluta entre ambos estados2 por lo que se desecharon estos procederes.
Los estudios del campo visual y la perimetría están indicados en pacientes con macroadenomas y con extensión supraselar del tumor o cuando se obtienen, al realizar el examen oftalmológico, síntomas y signos de afectación de esta esfera.14,28
En las embarazadas con hiperprolactinemia tumoral deben realizarse estudios evolutivos del campo visual para detectar un crecimiento del tumor.14,29
Así, en mujeres que presentan hiperprolactinemia, con todas las causas conocidas excluidas y estudios anatómicos normales, es planteable una hiperprolactinemia idiopática o funcional por lo que éste es solo un diagnóstico de exclusión.30
En conclusión, la causa más frecuente de hiperprolactinemia en la mujer es el adenoma hipofisario productor de prolactina (prolactinoma), los síntomas de más valor diagnóstico (amenorrea y galactorrea) están relacionados con el trastorno hormonal, aunque en casos de macroprolactinomas pueden presentarse manifestaciones neurooftalmológicas. Además de la determinación de prolactina, los estudios de mayor valor diagnóstico son los imagenológicos pues las pruebas dinámicas no son útiles. El estudio del campo visual es útil en el diagnóstico del macroprolactinoma y en la valoración evolutiva de los pacientes que lo padecen.
SUMMARY
A review of the most updated papers on hyperprolactinemia was conducted, taking into account that it is a common cause of amenorrhea, galactorrhea and infertility in women at reproductive age. Prolactin-producing adenomas are the most frequent tumors of hypophysis and microadenomas prevail among them. The symptoms of prolactinomas are more connected with hyperprolactinemia, but in cases of macroadenomas local symptoms as cephalalgia, defects of the field of vision, and more rarely, symptoms of compression of the cavernous sinus may appear. The dynamic function tests proposed for the differential diagnosis between the idiopathic and tumoral causes of hyperprolactinemia are not useful from the individual point of view. They could only be used for group discrimination. Once the diagnosis of hyperprolactinemia is confirmed, it is necessary to look for an injury compatible with a hypophyseal tumor through contrast computer tomography or nuclear magnetic resonance of the Turkish saddle. The objective of this theoretical review has been to show the main causes, clinical picture and the diagnostic procedures of this disease that is at present an important reason to attend the specialist's office.Subject headings: HYPERPROLACTINEMIA/etiology; HYPERPROLACTINEMIA/diagnosis.REFERENCIAS BIBLIOGRÁFICAS
- Vance ML, Thorner MO. Prolactin: hyperprolactinemic syndromes and management. En: Degroot LY, ed. Endocrinology. Philadelphia: W.B. Saunders, 1989:408-18.
- Andino N, Bidot C, Machado AJ. La prolactina y la infertilidad femenina. En: Padrón R. Temas de reproducción femenina. La Habana: Editorial Científico-Técnica, 1990:51-98.
- Archer DF. Current concepts of prolactin, physiology in normal and abnormal conditions. Fertil Steril 1977;28:125-34.
- Basil HY: Etiology and treatment of hyperprolactinemia. Semin Reprod Endocrinol 1992;10:228-30.
- Tadmor OP, Diamant I. Primary hypothyroidism presenting with amenorrhea, galactorrhea, hyperprolactinemia and enlarged pituitary. Harefuah 1992;122:76-80.
- Pietrobelli DJ. Asociación prolactinoma-hipotiroidismo primario. Neuroendocrinol Latinoam. 1990;(Extr):39--42.
- Pamir MN. Magnetic resonance imaging in the diagnosis of idiopathic giant-cells granulomatous hypophysitis a rare cause of hyperprolactinemia. Neurochirurgia (Stuttg) 1993;36:20-2.
- Palmer PE, Bogojavlensky S, Bhan AK, Scull RE. Prolactinoma in wall of ovarian dermoid cyst with hyperprolactinemia. Obstet Gynecol 1990;75:540-3.
- Hatfill SJ. Hyperprolactinemia in acute myeloid leukemia and indication of ectopic expression of human prolactin in blast cell a patient of subtype M4. Leuk Res 1990;14:57-9.
- Kira J. Hyperprolactinemia in multiple sclerosis. J Neurol Sci 1991;102:61-4.
- Buskila D. Improvement of psoriatic arthritis in a patient treated with bromocriptine for hyperprolactinemia. J Rheumatol 1991;18:611-4.
- Jara LJ, Gómez-Sánchez C, Silveira LH. Hyperprolactinemia in systemic lupus erythematosus: asociation with disease activity. Am J Med Sci 1992;303:222-6.
- Mahler C. Cushing's disease and hyperprolactinemia due to a mixed ACTH and prolactin secretion. Endocrinol Metabol 1981;53:863-6.
- Schlechte JA. Clinical impact of hyperprolactinaemia. Baillière´s. Clin Endocrinol Metab 1995;9:359-65.
- Fideleff H, Boquete HR, Orlandi AM, Wainstein L, Holland ME. Evolución de prolactinomas en adolescentes. Medicina (Buenos Aires) 1991;51:121-6.
- Cáceres O, Moloczni I, Artese R, Boero L, Driollet R, Rettori V. Síndrome de amenorrea primaria hiperprolactinémica con adenoma hipofisario. Aspectos clínicos y terapéuticos a propósito de cinco casos. Obstet Ginecol Latinoam 1990;8:117-24.
- Tyson D, Reggiard D, Sklar Ch, David R. Prolactin-secreting macroadenomas in adolescents. AJDC 1993;147:1057-61.
- Ciccarelli E, Camanni F. Diagnosis and drug therapy of prolactinoma. Drugs 1996;51:954-65.
- Strebel PM. Headache, hyperprolactinemia and prolactinomas. Obstet Gynecol 1986;68:195-8.
- Cunnah D, Besser GM. Management of prolactinomas. Clin Endocrinol 1991;34:231-5.
- Tonner MD, Schlechete J. Contemporary therapy of prolactin-secreting adenomas. Am J Med Sci 1993;306:395-7.
- Ando N. Prolactin disorders in patients with habitual abortion. Nippon Sanka Fujinka Gakkai Zasshi 1992;44:650-5.
- Jackson RD, Wortsman J, Malarkey WB. Characterization of a large molecule weight prolactin in women with idiopathic hyperprolactinemia and normal menses. J Clin Endocrinol Metabol 1985;61:258-61.
- Malarkey WB, Jackson R, Wortsman J. Long-term assessment of patient with macroprolactinemia. Fertil Steril 1988;50:413-8.
- Johnson MR, Hoare RD, Cox T, Dawson JM, Maccabe JJ, Huw Llewelyn D, et al. The evaluation of patients with a suspected pituitary microadenoma: computer tomography compared to magnetic resonance imaging. Clin Endocrinol 1992;36:335-8.
- Levy A, Lightman SL. Diagnosis and management of pituitary tumours. Br Med J 1994;308:1087-91.
- Moseley I. Computed tomography and magnetic resonance imaging of pituitary microadenoma. Clin Endocrinol 1992;36:333-4.
- Lesser RL, Zheutlin JD, Boghen D, Odel JG, Robbins RJ. Visual function improvement in patients with macroprolactinomas treated with bromocriptine. Am J Opthamol 1990;109:535-43.
- Prager D, Braunstein GD. Pituitary disorders during pregnancy. Endocrinol Metabol Clin North Am 1995;24:1-14.
- Coremblum B, Taylor PJ. Idiopathic hyperprolactinemia may include a distinct entity with a natural history different from that of prolactin adenomas. Fertil Steril 1988;49:544-6.
jueves, 9 de mayo de 2013
HIPERTIROIDISMO SUBCLINICO
Hipertiroidismo subclínico
Subclinical hyperthyroidism
Lisbet Rodríguez FernándezI; Marelys Yanes QuesadaII; Alina Acosta CedeñoI; Gilda Monteagudo PeñaIII; Abdel del BustoIV; Ana Margarita Montero MolinaIV
II Especialista de I Grado en Endocrinología. Asistente. Investigadora Agregada. INEN. La Habana, Cuba.
IIIEspecialista de II Grado en Endocrinología. Profesora e Investigadora Auxiliar. INEN. La Habana, Cuba.
IVResidente de Endocrinología. INEN. La Habana, Cuba.
RESUMEN
El hipertiroidismo subclínico se define por la presencia de niveles disminuidos o no detectables de tirotropina, asociados a concentración de tetrayodotironina y triyodotironina libres dentro de parámetros normales. Su prevalencia en la población varía entre un 0,5 y un 16 % aproximadamente, y es el tratamiento con levotiroxina sódica la causa más frecuente. No siempre resulta tan asintomático, y las afectaciones principales ocurren sobre el sistema cardiovascular y óseo. Esta condición médica puede ser reversible espontáneamente. Por lo controversial del tema, el presente trabajo trata los aspectos clínicos más relevantes y la conducta a seguir.
Palabras clave: Hipertiroidismo subclínico.
ABSTRACT
Subclinical hyperthyroidism is defined by presence of decreased o non-detected levels of thyrotropin, associated with free concentrations of tetraiodothyronine and triiodothyromime within normal parameters. Its prevalence in population differs between 0,5 % and 16 % approximately, and the sodium Levothyroxine treatment is the more frequent cause. Not always it is so asymptomatic, and main affections occur on cardiovascular and osseous system. This medical condition may be spontaneously reversible. Due to controversial of this topic, present paper approaching the more significant clinical features and the strategy to go on.
Key words: Subclinical hyperthyroidism.
INTRODUCCIÓN
El hipertiroidismo subclínico se caracteriza por la presencia de niveles disminuidos o no detectables de tirotropina (TSH) en relación con el rango de referencia establecido (0,3 a 3,75 mUI/L), asociados a la concentración de tetrayodotironina (T4) y triyodotironina (T3) libres dentro de parámetros normales en relación con el rango de referencia del laboratorio.1-3 La definición está basada, por tanto, en criterios de laboratorio y no clínicos. Con el desarrollo de ensayos clínicos de segunda y tercera generación para la determinación de TSH, este trastorno ha resultado ser relativamente frecuente en la práctica endocrinológica. Sin embargo, existen controversias respecto a la conducta a tomar con estos pacientes, que al diagnóstico, presentan pocas o ninguna manifestación clínica.1-3 El objetivo de este trabajo es brindar una panorámica sobre los aspectos clínicos, así como el tratamiento de este trastorno sobre el que no existe consenso internacional ni valores de corte de TSH que indiquen cuándo instaurar tratamiento específico, permitiendo una mejor comprensión y facilitando la conducta a seguir por los especialistas relacionados con el tema.
Esta situación bioquímica, tiene importancia por 3 razones fundamentales: la posibilidad de que ocurra una progresión a un hipertiroidismo bien establecido, los efectos cardiovasculares frecuentes en los individuos afectados y las consecuencias sobre el esqueleto.1-3 Estos cambios alteran la calidad de vida de los pacientes, sobre todo, en el adulto mayor, e incrementan el riesgo de morbilidad y mortalidad cardiovascular, así como la incidencia de fracturas óseas.1,4,5
Epidemiología
En general se estima que la prevalencia de hipertiroidismo en la población es de un 1 % aproximadamente, pero en el caso del hipertiroidismo subclínico no se conoce bien. En diferentes estudios publicados la prevalencia de esta condición médica varía entre un 0,5 y un 16 %.6,7 En Cuba no existen estudios estadísticos que reflejen su prevalencia. No hemos encontrado publicaciones realizadas en nuestro país relacionadas con este tema.
Etiología y diagnóstico diferencial
El hipertiroidismo subclínico es clasificado como endógeno en pacientes con producción de hormona tiroidea asociada a enfermedad de Graves-Basedow, bocio multinodular tóxico y adenoma tóxico, entre otras causas, y como exógeno, cuando la concentración disminuida o no detectable de tirotropina ocurre como resultado de la administración de hormonas tiroideas.1-3,8
La causa más frecuente es el tratamiento con levotiroxina sódica, y ocurre cuando la dosis empleada está por encima de las necesidades fisiológicas, en pacientes que tienen indicado este medicamento con el objetivo de sustituir o suprimir la función tiroidea. Con poca frecuencia puede observarse como consecuencia de la administración subrepticia de estos fármacos.1-3,9
Es importante señalar que en los adultos mayores el ritmo de aclaramiento plasmático de las hormonas tiroideas puede estar disminuido, y esto podría acompañarse de TSH disminuida sin manifestaciones clínicas de tirotoxicosis. Resulta necesario realizar el diagnóstico diferencial con la ingestión de algunos fármacos que pueden causar este trastorno (glucocorticoides, los agonistas de la somatostatina y la nandrolona). Otros medicamentos pueden producir aumento de la concentración de las hormonas tiroideas, como ocurre con la amiodarona o la ingestión de yodo en zonas de bocio endémico. La carbamazepina, la fenitoína y los salicilatos son capaces de desplazar las hormonas tiroideas de los sitios de unión a proteínas. En estos casos la realización de una historia clínica cuidadosa permitirá realizar un diagnóstico adecuado, y por tanto, tomar una conducta correcta.1-3,9,10
Manifestaciones clínicas
Existen varias investigaciones que reportan una disminución en la calidad de vida de los pacientes con manifestaciones de hiperfunción tiroidea subclínica. Los síntomas que pueden presentarse incluyen las palpitaciones, el temblor fino, la intolerancia al calor, las sudoraciones y el insomnio.10
Efectos sobre la esfera psíquica
La mayoría de los estudios realizados en pacientes con hipertiroidismo subclínico reportan efectos psicosomáticos, como la ansiedad, la sensación de temor, el nerviosismo, la hostilidad, la irritabilidad, la disminución de la concentración, y en algunos casos, sensación de bienestar.8,11-13 Se ha reportado, también, un incremento del riesgo de enfermedad de demencia y Alzheimer en pacientes mayores de 55 años.1,8,11
Efectos sobre la función cardiovascular
Las hormonas tiroideas tienen numerosos efectos sobre la función cardiovascular. La T3 regula la transcripción de genes que codifican la formación de proteínas contráctiles calcio dependientes en retículo sarcoplámico, determinantes en la contracción sistólica y la relajación diastólica. Por otro lado, la T3 provoca incremento en el tono simpático y descenso del tono parasimpático, unido a efectos directos sobre el músculo cardíaco. La mayoría de estas manifestaciones están relacionadas con cambios hemodinámicos, caracterizados por el aumento del gasto cardíaco como consecuencia del aumento de consumo de oxígeno por los tejidos, la disminución de la resistencia vascular sistémica y el incremento de la reabsorción renal de sodio, secundaria a la activación del eje renina-angiotensina-aldosterona, lo cual, unido al efecto directo de la T3 en la liberación de eritropoyetina, conlleva a un aumento del volumen de sangre y a un aumento de la contractilidad cardíaca.1,10,14-17
El hipertiroidismo subclínico produce a largo plazo efectos caracterizados por incremento de la masa ventricular izquierda, y como resultado, un trabajo cardíaco mayor. La fibrilación auricular y la insuficiencia cardíaca son las alteraciones cardiovasculares más frecuentes.16,17 La mayoría de los pacientes que desarrollan insuficiencia cardíaca durante el hipertiroidismo tienen una cardiopatía preexistente.17 Se ha reportado que el hipertiroidismo constituye el 15 % de todos los casos de fibrilación auricular de inicio reciente, y no importa si el origen de la levotiroxina sódica es exógeno o endógeno. La mayoría de los estudios muestran que la prevalencia de arritmias supraventriculares es alta en pacientes con hiperfunción subclínica. La fibrilación auricular es más frecuente en el adulto mayor. Hay estudios que reportan un aumento en la mortalidad cardiovascular en adultos mayores, a los 5 años de padecer el hipertiroidismo subclínico, asimismo se han publicado trabajos que reportan una mejoría clínica de los pacientes con esta condición que recibieron tratamiento para el hipertiroidismo. Otros efectos adversos del hipertiroidismo subclínico sobre la función cardiovascular incluyen alteraciones en la función diastólica y en la fracción de eyección ventricular en respuesta al ejercicio.4,5,8,10,14-17
Estudios retrospectivos describieron los resultados de estudios en pacientes con hipertiroidismo subclínico, uno de ellos reportó fibrilación auricular en un 28 % de pacientes con este trastorno respecto a un 10 % en sujetos controles eutiroideos. Otra investigación reportó una prevalencia de fibrilación auricular de 12,7 % contra 2,3 % en sujetos eutiroideos, de lo cual resultó un riesgo cardiovascular 5,2 (p<0 2005="" a.="" calidad="" con="" controles="" de="" del="" edad="" el="" en="" endocrinolog="" especialista="" espinosa="" eutiroideos="" farmacol="" gico.="" grado="" hipertiroidismo="" hueso="" ma.="" mediana="" mujeres="" nico="" optar="" para="" por="" primer="" que="" rce="" subcl="" sup="" superior="" t="" trabajo="" tulo="" veces="">1,18
Efectos sobre el esqueleto
El hipertiroidismo franco es un factor de riesgo de osteoporosis bien reconocido, pero los efectos del hipertiroidismo subclínico sobre la densidad mineral ósea no están bien definidos. Diferentes estudios indican que está relacionado con una disminución de la densidad mineral ósea.1,3 Se considera que esta situación es un factor de riesgo de osteoporosis, especialmente en la mujer posmenopáusica. Existen estudios que han reportado una disminución de la densidad ósea, estadísticamente significativa, y asociada a manifestaciones clínicas, sobre todo, en el cuello femoral y el radio de los pacientes estudiados, respecto a controles.8 También se ha reportado la mejoría en la densidad mineral ósea en mujeres posmenopáusicas con hipertiroidismo subclínico, posterior al restablecimiento de la concentración de tirotropina dentro del rango normal.1,3,8,10
En el año 2005 se realizó una investigación en el INEN, que reportó una mayor frecuencia de mala calidad ósea en mujeres de edad mediana que recibían tratamiento con hormonas tiroideas en dosis suprafisiológicas.19-21
Tratamiento
Los pacientes que presentan hipertiroidismo subclínico y están asintomáticos deben tener seguimiento clínico y por laboratorio, teniendo en cuenta la alta frecuencia (alrededor de un 50 %) en que ocurre una remisión espontánea, por lo que el tratamiento podría ser innecesario en múltiples situaciones.2,3,10
El panel de expertos NHANES III ha recomendado no realizar estudios de cribado, y que debe dosificarse la TSH solo en situaciones clínicas que sugieran diagnóstico de hiperfunción tiroidea.3,10 No obstante, por lo general, estos pacientes desde el inicio presentan alguna manifestación clínica que motivó el estudio de la función tiroidea y que permitió detectar esta situación médica, por lo que la conducta definitiva se toma casi siempre en un período no mayor de un año.
Si el paciente toma alguno de los fármacos relacionados con hipertiroidismo subclínico y tiene sintomatología, de confirmarse que la TSH está disminuida, deberá suspenderse o sustituirse el medicamento empleado. Aquellos pacientes que tengan manifestaciones clínicas, sufran arritmias supraventriculares o presenten pérdida de peso sin otra explicación, podrían ser candidatos a tratamiento con antitiroideos de síntesis o yodo radiactivo.2,3,8,10,20,21
En los pacientes que reciben tratamiento sustitutivo con levotiroxina sódica, la dosis debe ajustarse de forma tal que la concentración sérica de TSH se mantenga dentro del rango de referencia normal, excepto en los pacientes ancianos, en los que el objetivo principal del tratamiento del hipotiroidismo es lograr una mejoría considerable de la calidad de vida del paciente, y no la normalización de la TSH sérica.4,5 La presencia de hipertiroidismo subclínico en estos pacientes aumenta el riesgo de presentar fibrilación auricular 3 veces más respecto a adultos jóvenes, por lo tanto, debe administrarse la mínima dosis de levotiroxina sódica que logre el bienestar del paciente. En general, es importante el monitoreo periódico de la TSH en los pacientes con hipotiroidismo primario con tratamiento sustitutivo de levotiroxina sódica, lo que pudiera realizarse anualmente, una vez que se haya logrado el control de la enfermedad.2,3,8,10,20,21
Actualmente el tratamiento con dosis supresiva de levotiroxina sódica está indicado posterior a la tiroidectomía total en pacientes con carcinoma diferenciado de tiroides, excepción en la que podría ser conveniente mantener una supresión de la TSH sérica. Sin embargo, muchos pacientes no llegan a tolerar estas dosis porque aparecen manifestaciones claras de tirotoxicosis, por lo que el médico de asistencia se ve obligado a la disminución en la dosis de levotiroxina sódica.2,8,10
La otra indicación de dosis supresiva de levotiroxina sódica es el tratamiento del bocio eutiroideo y del nódulo único del tiroides, y su uso tiene como objetivo evitar el crecimiento de la glándula, pero debido a los efectos indeseables que puede ocasionar esta conducta, es preferible el empleo de dosis que logren una supresión mínima de la TSH. El tratamiento podría indicarse por un período de 3 a 6 meses, y después el especialista deberá reevaluar al paciente y valorar si mantenerlo o suspenderlo. Deben considerarse algunas situaciones en el momento de decidir si tratar o no el bocio eutiroideo: los pacientes con bocios pequeños y asintomáticos pueden ser evaluados periódicamente desde el punto de vista clínico y ultrasonográfico. El crecimiento del bocio es variable, y algunos pacientes tienen bocios con tamaño estable durante muchos años sin necesidad de tratamiento. Los bocios nodulares tienen poca respuesta al tratamiento respecto a los bocios difusos, por lo que el empleo de hormonas tiroideas, por lo general, resulta ineficaz. Los pacientes jóvenes con nódulos sólidos pequeños o recién diagnosticados tienen una mejor respuesta al tratamiento supresivo con levotiroxina sódica, respecto a otros subgrupos de pacientes.10,20,21
Se concluye que el hipertiroidismo subclínico no siempre es asintomático, y repercute con mayor frecuencia sobre el sistema cardiovascular y óseo. Las manifestaciones clínicas debidas a hipertiroidismo subclínico pueden ser reversibles, si se logra mantener la concentración de TSH dentro del rango de referencia normal. El 50 % de los pacientes con hipertiroidismo subclínico pueden evolucionar a una normalización de la función tiroidea espontáneamente.
REFERENCIAS BIBLIOGRÁFICAS
1. Biondi B, Palmieri EA, Klain M, Schlumberger M, Filetti S, Lombarda G. Subclinical hyperthyroidism: clinical features and treatment options. European Journal of Endocrinology. 2005;152(1):1-9.
2. Galofré JC. Manejo del hipertiroidismo subclínico. Rev Med Univ Navarra. 2007;51(1):18-22.
2. AACE. Thyroid Guidelines. Subclinical Hyperthyroidism. Endocr Pract. 2002;8(6):457-69.
3. Ayala C, Cózar MV, Rodríguez JR, Silva H, Pereira JL, García-Luna PP. Enfermedad tiroidea subclínica en la población anciana sana institucionalizada. Med Clin (Barc). 2001;117(1):534-5.
4. Real JT, Ascaso JF. Hipertiroidismo en el anciano. Med Clin (Barc). 2002;118(20):784-7.
5. McDermott MT, Woodmansee WW, Haugen BR, Smart AE, Ridgway CH. The Management of Subclinical Hyperthyroidism by Thyroid Specialists. Thyroid. 2003;13(12):1133-9.
6. Flynn RWV, MacDonald TM, Morris AD, Jung RT, Leese GP. The Thyroid Epidemiology, Audit, and Research Study: Thyroid Dysfunction in the General Population. The Journal of Clinical Endocrinology & Metabolism. 2004;89(8):3879-84.
7. Toft AD. Subclinical Hyperthyroidism. N Engl J Med. 2001;345(7):512-6.
8. Charkes ND. The many causes of subclinical hyperthyroidism. Thyroid. 1996;6(5):391-6.
9. Terry F, Davies P, Reed Larsen. Thyrotoxicosis. En: Larsen PR, Kronemberg HM, Melmed S, Polonsky KS. Williams textbook of Endocrinology. 11th. Philadelphia: Editorial Elsevier; 2008.p.333-7.
10. Stott DJ, McLellan AR, Finlayson J, Chu P, Alexander WD. Elderly patients with suppressed serum TSH but normal free thyroid hormone levels usually have mild thyroid overactivity and are at risk of developing overt hyperthyroidism. Quarterly Journal of Medicine. 1991;78:77-84.
11. Schlote B, Schaaf L, Schmidt R, Pohl T, Vardarli I, Schiebeler H, et al. Mental and physical state in subclinical hyperthyroidism: investigations in a normal working population. Biological Psychiatry. 1992;32:48-56.
12. Kalmijn S, Mehta KM, Pols HA, Hofman A, Drexhage HA, Breteler MM. Subclinical hyperthyroidism and the risk of dementia. The Rotterdam study. Clinical Endocrinology. 2000;53:733-7.
13. Gullu S, Altuntas F, Dincer I, Erol C, Kamel N. Effects of TSH-suppressive therapy on cardiac morphology and function: beneficial effects of the addition of b-blockade on diastolic dysfunction. European Journal of Endocrinology. 2004;150:655-61.
14. Klein I, Ojamaa K. Thyroid Hormone and The Cardiovascular System. N Engl J Med. 2001;344(7):222-35.
15. Federico Moreno K, Paoli de Valeri M, Macchioli RO, Núñez Medina TJ, Arata Bellabarba G. Hipertiroidismo subclínico exógeno: efecto en el sistema cardiovascular. Rev Esp Endocr Nutr. 2008;55(6):243-8.
16. Fazio S, Palmieri EA, Lombardi G, Biondi B. Effects of Thyroid Hormone on the Cardiovascular System. Recent Progress in Hormone Research. 2004;59(2):31-50.
17. Auer JA, Scheibner P, Mische T, Langsteger W, Eber O, Eber B. Subclinical hyperthyroidism as a risk factor for atrial fibrillation. American Heart Journal. 2001;142:838-842.
18. Tenerz A, Forberg R, Jansson R. Is a more active attitude warranted in patients with subclinical thyrotoxicosis? Journal of Internal Medicine. 1999;228:229-33.
19. Surks MI, Ortiz E, Daniels GH, Sawin CT, Col NF, Cobin RH, et al. Subclinical thyroid disease: scientific review and guidelines for diagnosis and management. JAMA. 2004;291(2):228-38.
20. Hoogendoorn E, den Heijer M, van Dijk A, Hermus A. Hipertiroidismo Subclínico: ¿Tratarlo o no? Postgraduate Medical Journal. 2004;80(945):394-8.
Recibido: 21 de febrero de 2009.Aprobado: 30 de marzo de 2009.
Lisbet Rodríguez Fernández. Instituto Nacional de Endocrinología. Calle Zapata y D, Vedado, municipio Plaza, Ciudad de La Habana, Cuba.
martes, 30 de abril de 2013
VASCULITIS
Vasculitis
1.Introducción
Las vasculitis comprenden un grupo heterogéneo de entidades relativamente frecuentes (suponen 1 de cada 300 ingresos en un hospital universitario) de etiologías y manifestaciones diversas, que se caracterizan por la inflamación de los vasos sanguíneos, arterias, venas o ambos, lo cual compromete su función con el desarrollo de isquemia y necrosis.
La inflamación vascular puede acompañarse de sintomatología general (fiebre, astenia, afectación del estado general) y/o el desarrollo de manifestaciones locales orgánicas dependientes del órgano afecto por la vasculitis (afectación cutánea, síntomas neurológicos, dolor abdominal, compromiso renal, etc.). La piel y el tejido subcutáneo se afectan frecuentemente en las vasculitis. Esta alta frecuencia de afectación es probablemente debida a diversos factores incluyendo el gran número de vasos dermicos, la exposición al frio y la presencia de fenómenos de estasis vascular que favorecerían el desarrollo de vasculitis con afectación cutánea. Las vasculitis pueden manifestarse a nivel cutáneo de diversas formas, desde cambios en la coloración, edema, púrpura, equímosis y necrosis (úlceras), siendo la manifestación más frecuente el desarrollo de púrpura palpable en extremidades inferiores. El desarrollo de una púrpura palpable puede ser la manifestación de una vasculitis benigna de corta duración, puramente cutánea a una manifestación cutánea de una vasculitis sistémica que puede acompañarse de afectación orgánica con compromiso vital. La elevada frecuencia de manifestaciones cutáneas de las vasculitis hace que el reconocimiento de estas lesiones, sea importante para el diagnóstico de las mismas.
2.Patogénesis
El diagnóstico y la clasificación de las vasculitis se basa especialmente en los mecanismos patogénicos que las producen. Se pueden clasificar las vasculitis en base a los mecanismos de producción que incluyen: la infección directa de los vasos, mecanismos inmunes y vasculitis de causa desconocida (Tabla 1). La mayor parte de vasculitis pueden catalogarse dentro de las causas infecciosas o en las inmunológicas. Dado que el tratamiento de las vasculitis infecciosas es radicalmente diferente del de las vasculitis mediadas por daño inmunológico, es importante realizar la distinción entre las dos formas de vasculitis en las fases iniciales de la valoración de estos enfermos. Es importante descartar una causa infecciosa antes de instaurar un tratamiento inmunosupresor. La mayoría de las vasculitis están mediadas por mecanismos inmunes, y se clasifican según los 4 tipos de reacción de hipersensibilidad de Gell y Coombs:
Tabla 1 Mecanismos patogénicos de las vasculitis
|
|
- Tipo I (vasculitis alérgica o anafiláctica: incluye las vasculitis asociadas a estados atópicos, urticaria vasculitis y síndrome de Churg-Strauss. Se caracterizan por la presencia de niveles séricos y tisulares de IgE elevados. En la fase vasculítica se caracteriza por infiltración angiocéntrica de los vasos por eosinófilos.
- Tipo II (citotóxica o citolítica):
- vasculitis mediadas por ANCA (granulomatosis de Wegener, poliangeitis microscópica y síndrome de Churg-strauss). Los ANCA (anticuerpos anticitoplasma de neutrófilos) son capaces de activar los neutrófilos y las células endoteliales, así como inducir la apoptosis acelerada de los neutrófilos.
- Anticuerpos anti-células endoteliales endoteliales (AECA). Los AECA pueden causar vascultis por daño directo o por activación del complemento, están involucrados en la enfermedad de Behçet y la enfermedad de Takayasu y tienen especificidad por diferentes regiones vasculares, afectando a vasos de pequeño tamaño en la enfermedad de Behçet y vasos de gran tamaño en la enfermedad de Takayasu.
- Tipo III (mediada por inmunocomplejos): el depósito de inmunocomplejos da lugar a la activación del complemento y liberación de los componentes C3 y C5, que producen quimiotáxis de neutrófilos y liberación de enzimas proteolíticas que dañan la pared vascular. Es el grupo más amplio de vasculitis entre las que encontramos la vasculitis leucocitoclástica cutánea, el síndrome de Schonlein-Henoch y poliarteritis nodosa, entre otras formas.
- Tipo IV (citotóxica) vasculitis mediada por linfocitos T: en este grupo se incluyen aquellas vasculitis granulomatosas que se caracterizan por la presencia en la pared de los vasos de infiltrados inducidos por linfocitos T, especialmente Th1, que serían responsables por medio de la producción de interferon-γ, de la acumulación de macrófagos que fagocitarían las fibras elásticas. En este grupo de vasculitis se encuentra la arteritis de la temporal.
Las vasculitis son un grupo de síndromes heterogéneo que sin embargo comparten varias manifestaciones entre ellas. No existe un esquema de clasificación ideal de las vasculitis. Se pueden clasificar en relación a si
- son primarias o secundarias (tabla 2),
- en relación al tamaño del vaso afecto y las características histológicas encontradas en la histología (tabla 3 ) o
- en base a los mecanismos patogénicos (tabla 4).
4.Manifestaciones cutáneas de vasculitis
4.1. Clinica: Las manifestaciones clínicas cutáneas de las vasculitis en la piel orientan hacia el diagnóstico de vasculitis, pero no son especificas de ninguna entidad específica. Las lesiones cutáneas son útiles como signo diagnóstico y su estudio histológico y mediante inmunofluorescencia directa sirven para obtener confirmación diagnóstica de vasculitis. Las manifestaciones más características son el desarrollo de púrpura palpable y de nódulos, pero pueden observarse otras manifestaciones cutáneas tales como petequias, equimosis, máculas eritematosas, lesiones de urticaria, livedo reticularis, necrosis, ulceras, vesículas, pústulas, ampollas, lesiones a tipo pioderma gangrenoso, lesiones tipo eritema nodoso y lesiones a tipo síndrome de Sweet. El predominio de una lesión clínica u otra vendrá determinado por la localización del vaso afecto y por las características del proceso inflamatorio.
Las vasculitis con afectación de vasos de pequeño calibre en la piel se manifiestan principalmente por la púrpura, que con frecuencia es palpable y afecta principalmente a extremidades inferiores (debido a la presión hidrostática). La púrpura suele desarrollarse en brotes sucesivos, inicialmente son máculas de coloración rojiza, que evolucionan hacia placas y pápulas, que pueden ser desde unos milímetros hasta varios centímetros de diámetro. Las lesiones más grandes son mas equimóticas que purpúricas. El color puede evolucionar desde el rojo-purpúrico hasta parduzco, en relación a la evolución de la degradación de la sangre extravasada. En algunos pacientes, especialmente aquellos en los que la vasculitis mediada por inmunocomplejos se acompaña de una gran activación de complemento pueden dar lugar a focos de edema cutáneo que se manifiesta clínicamente por brotes de urticaria, que generalmente dura mas de 24 horas y evoluciona hacia lesiones purpúricas. Los nódulos suelen ser calientes, tumefactos y rojos y pueden estar rodeados por lesiones de livedo reticulares. Los nódulos cutáneos se observan en las vasculitis que afectan a vasos de mayor calibre, como la poliarteritis nodosa - cutánea o sistémica-, la granulomatosis de Wegener, el síndrome de Churg-Strauss y la arteritis de células gigantes.
Tabla 6 Manifestaciones cutáneas de las vasculitis
4.2 Histología: El hallazgo histológico más frecuente en las vasculitis es el de es una vasculitis neutrofílica leucocitoclastica , que se caracteriza por la presencia de infiltrado inflamatorio afectando a la pared vascular con presencia de edema endotelial, infiltrado inflamatorio con predominio de polimorfonucleares neutrófilos, leucocitoclasia (degranulación y fragmentación de los polimorfonucleares dando lugar al polvo nuclear) hemorragia y trombosis. El desarrollo de nódulos cutáneos en pacientes con vasculitis pueden observarse en los casos en que existe un inflamación cutánea o subcutánea centrada en un vaso. La realización de estudios de inmunofluorescencia directa son útiles en el diagnóstico de vasculitis ya que nos permiten la demostración de inmunoclomplejos depositados en los vasos dérmicos y saber por que inmunoglobulina están constituidos (IgG, IgM, IgA, C3 y/o fibrinogeno).
5.Síndromes vasculíticos
Las vasculitis que nos van a interesar más son las vasculitis sistémicas primarias producidas por trastornos inmunes como la vasculitis o angeitis leucocitoclástica cutánea, las vasculitis sistemicas primarias asociadas a ANCAS incluyendo la granulomatosis de Wegener, la granulomatosis de Churg-Straus y la poliangeitis microscopica, asi como las vasculitis sistemicas con afectación de vasos de gran tamaño como arteritis temporal y la poliarteritis nodosa.
5.1.Vasculitis por hipersensibilidad o vasculitis leucocitoclastica cutánea
El término de vasculitis leucocitoclastica o necrotizante cutánea engloba un grupo amplio y heterogéneo de síndromes que se caracterizan por la inflamación, mediada por inmunocomplejos, de vasos capilares, vénulas y ocasionalmente arteriolas cutáneas, con cambios histológicos que se describen bajo el término de vasculitis leucocitoclástica (edema endotelial, infiltración por polimorfonucleares, cariorexis, hemorragia y trombosis).Incidencia: La vasculitis por hipersensibilidad es la forma más frecuente de vasculitis, representando entre el 17-29% de los casos de vasculitis. Puede afectar a cualquier edad, el 10% de los afectos son niños. Clínica: Las manifestaciones clínicas más frecuentes son dermatológicas. Más del 95% de los pacientes desarrollan púrpura palpable. Otras manifestaciones clínicas incluyen urticaria, eritema multiforme y livedo reticularis. Las lesiones cutáneas suelen tener una distribución simétrica afectando a áreas acras y de declive, inicialmente pueden no ser purpúricas y en su progresión desde pequeñas pápulas purpúricas pueden aumentar de tamaño y evolucionar hasta formar placas de varios centímetros y desarrollar vesículas y ulceración.
Etiología: El mecanismo principal de producción es el depósito de inmunocomplejos. Estos inmunocomplejos pueden activar el sistema de complemento, produciendo la fracción C3 y C5, lo que produce la quimiotaxis de polimorfonucleares, los cuales liberan enzimas lisosomales que producen el daño tisular. El origen de los complejos Ag-Ac es idiopática en el 50% de los casos. Un 20% se asocian a infecciones especialmente víricas y bacterianas, un 22% se asocian a medicaciones y un porcentaje menor a enfermedades del tejido conectivo (12%) y a antigenos tumorales (<5 br="" de="" especialmente="" linfoproliferativo="" mieloproliferativo.="" nbsp="" o="" origen="">Histologia: El estudio histológico de las lesiones de púrpura palpable demuestra una vasculitis leucocitoclástica que consiste en la presencia de inflamación centrada en un vaso con edema endotelial, necrosis fibrinoide de los vasos capilares, e infiltrado inflamatorio de predominio polimorfonuclear, con fragmentación de los núcleos (leucocitoclasia), hemorragia y trombosis. Los estudios de inmunofluorescencia directa pueden demostrar la presencia de inmunoglobulinas, complemento y firbrinogeno en los vasos dérmicos.
Subgrupos de vasculitis leucocitoclástica cutánea: Existen varias formas clínicas de vasculitis leucocitoclástica cutánea que se pueden agrupar separadamente y que están resumidas en la tabla 10.
- Síndrome de Henoch-Schonlein: Es la forma más frecuente de vasculitis en niños. Se caracteriza por el desarrollo de rash purpúrico, artralgia/artritis, afectación gastrointestinal y nefritis. Las lesiones clínicas son polimorfas las lesiones más características son de púrpura palpable, pero pueden desarrollar pápulas, urticaria, angioedema, vesículas, necrosis y livedo reticulares. Las lesiones afectan principalmente a miembros inferiores y glúteos. Las lesiones cursan a brotes. Ocasionalmente pueden acompañarse de fiebre, malestar, artralgias o mialgias. En general suele reservarse la clasificación en este síndrome para aquellos casos en que se demuestra la presencia de inmunocomplejos circulantes de clase IgA o la presencia de depósitos inmunes en piel de clase IgA
- Urticaria vasculitis: se caracteriza por el desarollo de lesiones cutáneas en forma de habón que a diferencia de lo que ocurre en la urticaria aguda tienen a perdurar con duración de más de 24 horas, deja pigmentación residual, generalmente se asocia con sensación de quemazon más que con picor, suele asociar síntomas sistémicos como fiebre, angioedema, artralgias y artritis y dolor abdomina. Hasta un 30% de pacientes asocian hipocomplementemia y su presencia se asocia con una mayor gravedad.
- Vasculitis crioglobulinemica: Las crioglobulinas son inmunoglobulinas que precipitan con el frio y que producen daño orgánico por dos mecanismos principales, la oclusión vascular (síndrome de hiperviscosidad, principalmente en la crioglobulinemia tipo I y por mecanismos inmunes principalmente en las crioglobulinemias mixtas. Existen 3 tipos básicos de crioglobulinemias, el tipo I monoclonal IgM o IgG, el tipo II mixta monoclonal IgM y polilclonal IgG y el tipo III mixta policlonal IgG e IgM. Las crioglobulinas se relacionan con diversas patologías que incluyen infecciones, enfermedades autoinmunes y neoplasias siendo la más frecuente la infección por virus de la hepatitis C. El porcentaje de pacientes con crioglobulinemia que desarrolla lesiones clínicas varía desde el 2 al 50% de los pacientes, siendo los hallazgos clínicos más característicos la triada de púrpura, artralgias y debilidad que son las manifestaciones debutantes en el 80% de los pacientes, siendo la púrpura la manifestación más característica. Otras manifestaciones cutáneas incluyen úlceras, generalmenet alrededor del maleolo y lesiones isquémicas. El desarrollo de úlceras cutáneas y lesiones de gangrena digital empeoran el pronóstico con un mayor riesgo de infección, sepsis y muerte.
5.2. Vasculitis sistémica primaria asociadas a ANCA
Aproximadamente un 5% de pacientes que se presentan con vasculitis cutánea tienen una vasculitis sistémica y alrededor de un 3% tienen una vasculitis sistémica asociada a ANCA. Estas formas de vasculitis consitutuyen un grupo de enfermedades afectando a vasos de pequeño y mediano calibre que incluyen la granulomatosis de Wegener, la poliangeitis microscópica y el Síndrome de Churg-Strauss. Los ANCA son anticuerpos dirigidos contra antígenos de los polimorfonucleares. Existen 2 patrones de ANCAS : el patrón citoplasmático que incluye los anticuerpos contra la proteinasa 3, y el patron periférico dirigido contra la mieloperoxidasa. En la actualidad se piensa que ciertas moléculas proinflamatorias como el TNF-α y la IL-1 inducen la translocación de la proteinasa 3 y la mieloperoxidasa hacia la superficie de los neutrófilos. Estos antígenos se unen a los ANCA, activando a los neutrofilos y aumentando su adherencia a las células endoteliales dando lugar al daño vascular.
- granular citoplásmico (C-ANCA, PR3-ANCA, con especificidad ante el antígeno citoplásmico proteinasa 3)
- patrón perinuclear (p-ANCA, MPO-ANCA, con especificidad contra el antígeno mieloperoxidasa)
5.2.1.Granulomatosis de Wegener (GW): La granulomatosis de Wegener es una vasculitis sistémica primaria caracterizada por la triada clínica de vasculitis granulomatosa del tracto superior e inferior, glomerulonefritis y grados variables de vasculitis de pequeño vaso. Los pacientes con GW generalmente presentan síntomas de vías respiratorias superiores, incluyendo sinusitis, obstrucción y perforación nasal que puede dar lugar a una deformidad en silla de montar. Otros síntomas frecuentes incluyen otitis media, dolor de oído y disminución de la capacidad auditiva. La afectación pulmonar puede manifestarse en forma de tos productiva y hemoptisis o puede ser asintomática. La afectación renal en forma de glomerulonefritis determina el pronóstico de los enfermos. Al menos el 50% de los pacientes tienen afectación mucocutánea que puede ser la forma de presentación hasta en un 12% de casos. Las manifestaciones cutáneas pueden ser de 3 tipos: 1)púrpura palpable como manifestación de una vasculitis leucocitoclástica de pequeño vaso, 2) nódulos subcutáneos y úlceras como manifestación de una vasculitis de mediano vaso y 3) lesiones polimorfas que incluyen pápulas y nódulos necróticos en las áreas periarticulares, úlceras a tipo pioderma gangrenoso y lesiones de hiperplasia gingival granulomatosa. La afectación cutánea en la granulomatosis de Wegener se asocia con afectación sistémica activa y progresiva. Los criterios para el diagnóstico de la GW están resumidos en la tabla 8. La granulomatosis de Wegener se asocia de forma específica con la presencia de ANCA con especificidad contra la proteinasa 3, con patrón citoplásmico. en los pacientes con enfermedad activa tiene una alta sensibilidad y especifidad.
5.2.2.Poliangeitis microscópica: Forma de vasculitis de pequeño vaso que se asocia frecuentemente a enfermedad renal rápidamente progresiva (glomerulonefritis necrotizante segmentaria focal), afectación cutánea (>75% púrpura palpable), presencia de anticuerpos contra p-ANCA, en ausencia de granulomas extravasculares y depósitos de inmunocomplejos en la inmunofluorescencia directa. La poliangeitis microscopica tiene un pronostico peor que las otras formas de vasculitis asociadas a ANCAS con una supervivencia a los 5 años del 45%. .
5.2.3.Vasculitis granulomatosa y alérgica de Churg-Strauss: La enfermedad de Churg-Strauss es una vasculitis sistémica primaria rara que se observa en pacientes con asma. Se caracteriza por afectar a vasos de tamaños variables con formación de granulomas intra y extravasculares -con intensa presencia de eosinófilos en el infiltrado- y por afectar a pacientes con historia de asma, atopia y eosinofilia periférica. El síndrome de Churg-strauss está considerado resultado de una reacción de hipersensibilidad tipo I, en el cual la proliferación de linfocitos CD4+, TH2 estimulada por diversos alergenos -inhalados, vacunas, medicaciones o infecciones. Los linfocitos TH2 producen interleucina 4, 5 y 13 que estimulan la acumulación de mastocitos, basófilos y especialmente eosinófilos que producen el daño tisular.
Suele asociar sintomatología sistémica con fiebre, mal estado general y pérdida de peso. La manifestación clínica principal e inicial es el desarrollo de asma, que suele afectar a personas con antecedentes de atopia (bronquitis asmática, rinitis o conjuntivitis alérgica y dermatitis). En un segundo estadio las crisis asmáticas se agravan y suelen acompañarse de infiltrados pulmonares con alteraciones radiológicas y eosinofilia periférica y en un tercer estadio se desarrollan signos y síntomas propios de una vasculitis, solo en este último estadio es posible realizar el diagnóstico del síndrome. El 50% de los pacientes fallecen por afectación cardíaca de la enfermedad. El 70% de los pacientes desarrollanlesiones cutáneas en forma de nódulos cutáneos y subcutáneos, denominados granulomas extravasculares de Churg-Strauss, que pueden observarse tanto en la vasculitis de Churg Strauss como en otras enfermedades reumáticas. Otras lesiones cutáneas incluyen el desarrollo de púrpura palpable y livedo reticularis. El 75% de los pacientes desarrollan manifestaciones neurológicas. La analítica más característica muestra una eosinofília periférica con valores de entre 5 y 10x109 eosinófilos/L. El nivel de eosinofilia es un buen marcador para el tratamiento de la enfermedad. El diagnostico de vasculitis puede sugerirse ante la presencia de asma, eosinofilia, infiltrados pulmonares y datos de afectación multisistémica en pacientes con antecedentes atópicos. Los criterios diagnósticos del Síndrome de Churg-Strauss están resumidos en la tabla 9.(ver caso NEJM )
5.3.Arteritis temporal (Vasculitis de células gigantes): La arteritis temporal es la forma de vasculitis sistémica primaria de causa desconocida que afecta a arterias de calibre medio y gran calibre afectando especialmente a las ramas extracraneales de la carótida. Afecta especialmente a personas mayores de 50 años. Cursa con afectación segmentaria de los vasos, con presencia de un infiltrado mononuclear -linfocítico y histiocítico- afectando especialmente a la capa media de las arterias. Se clasifica dentro de las vasculitis mediadas por linfocitos T, observándose en los vasos un infiltrado de predomino CD4, Th1. Clínicamente se caracteriza por síntomas asociados al proceso inflamatorio sistémico (fiebre, malestar general, anorexia, depresion) y manifestaciones clínicas que reflejan la isquemia tisular debida a la estenosis del vaso (cefalea, ceguera, tumefacción y ulceración del cuero cabelludo, claudicación mandibular). Más de 2/3 de los pacientes tienen cefalea que es también el sintoma inicial más frecuente, esta cefalea suele ser de inicio brusco, intensa y de predominio temporal. El examen físico puede mostrar una arteria temporal que se puede visualizar con facilidad en forma de un cordón, doloroso y nodular con el pulso disminuido o ausente . Los criterios diagnósticos están resumidos en la tabla 6. Un 50% de los pacientes asocia polimialgia reumática, síndrome estrechamente relacionado con la arteritis temporal y caracterizado por mialgias, tumefacción, dolor articular, fiebre y pérdida de peso. Las lesiones cutáneas son raras y consisten en el desarrollo de úlceras necróticas en el territorio de la temporal y se asocian a una marcada estenosis de la luz vascular. El diagnóstico debe realizarse en base a los hallazgos clínicos y confirmarse mediante la biopsia de la arteria temporal. La imagen histológica clásica se observa en el 50% de los pacientes y muestra un infiltrado granulomatoso de las capas media e intima de la arteria temporal. Otro 50% de pacientes no muestra los hallazgos característicos, observándose una panarteritis con un infiltrado inflamatorio mixto, con presencia de células mononucleadas y escasos neutrófilos y eosinófilos. Una vez sospechado el diagnóstico debe iniciarse el tratamiento de forma precoz para evitar las complicaciones oculares (la pérdida completa o parcial de la visión de uno o ambos ojos afecta al 20% de los pacientes y es un síntoma presente en las fases iniciales de la enfermedad, -un 44% presentan episodios de amaurosis fugax). La buena y rápida respuesta al tratamiento se considera también criterio diagnóstico de esta entidad.
5.4.Poliarteritis nodosa (PAN): La poliarteritis nodosa es una vasculitis sistémica primaria en la que existe una vasculitis necrotizante de arterias de pequeño y mediano calibre, con marcada afectación renal y visceral. La afectación es segmentaria y tiene predilección por las zonas de bifurcación vascular. Afecta principalmente a varones de mediana edad. Con frecuencia asocia manifestaciones sistémicas generales como fiebre, mal estado general y pérdida de peso. El 70% tienen afectación renal (en forma de arteritis y/o glomerulonefritis, siendo esta la principal causa de muerte), el 60% tienen afectación neurológica periférica con un patrón de afectación de mononeuritis múltiple o polineuropatía. El 50% tienen afectación cutánea incluyendo el desarrollo de nódulos, úlceras y livedo reticularis. En la PAN no existe afectación de las arterias pulmonares. El origen de la PAN es desconocido, se considera dentro de las vasculitis mediadas por inmunocomplejos (reacción tipo III). Se han detectado antígenos de la hepatitis B hasta en un 50% de los pacientes. El hallazgo de inmunocomplejos depositados a nivel de la pared vascular diferencia la PAN de la poliangeitis microscópica. Los cambios histológicos consisten en una inflamación transmural pleomorfica con polimorfonucleares, necrosis fibrinoide, hemorragia y formación de aneurismas. Los criterios diagnósticos de la PAN están resumidos en la tabla 7.
Existe una forma de poliarteritis nodosa limitada a la piel, poliarteritis nodosa cutánea , de curso benigno, donde la afectación visceral está ausente, siendo las manifestaciones más frecuentes el desarrollo de nódulos cutáneos, livedo reticularis y ulceración cutánea.
Existe una forma de poliarteritis nodosa limitada a la piel, poliarteritis nodosa cutánea , de curso benigno, donde la afectación visceral está ausente, siendo las manifestaciones más frecuentes el desarrollo de nódulos cutáneos, livedo reticularis y ulceración cutánea.
6.Actitud diagnóstica ante una vasculitis con manifestaciones cutáneas
Con frecuencia los pacientes con vasculitis debutan con lesiones cutáneas, especialmente púrpura palpable. Las manifestaciones clínicas y patológicas de las vasculitis en piel no son específicas para ningún tipo de vasculitis, por ejemplo, la púrpura palpable que es resultado de la inflamación de un vaso en dermis puede ser originada por una vasculitis infecciosa (pe vasculitis por neiseria), por una vasculitis mediada por inmunoclomplejos ( vgr: la enfermedad del suero, las vasculitis por crioglobulinemia, o la púrpura de Henoch Schonlein), por una vasculitis asociada a ANCA ( granulomatosis de Wegener, poliangeritis microscópica,), una vasculitis alérgica (por una reacción medicamentosa) una vasculitis asociada a un enfermedad reumática (vgr: lupus eritematoso, arteritis reumatoide y síndrome de Sjogren) . De la misma manera, los nódulos cutáneos y subcutáneos inflamatorios pueden ser originados por una variedad de vasculitis, incluyendo la poliarteritis nodosa, la poliangitis microscópica, la granulomatosis de Wegener, el síndrome de Churg-strauss. Cuando existe la sospecha clínica de que estamos ante una vasculitis los pacientes deben ser estudiados de forma minuciosa para determinar la etiología de la vasculitis, la extensión de la afectación vascular y para establecer el tratamiento adecuado.Los estudios clínicos a realizar cuando se sospecha el diagnóstico de vasculitis cutánea deben estar dirigidos de la siguiente manera (Figura 1). Se han descrito diversos algoritmos diagnósticos que deben ser aplicados en grupos grandes de pacientes para comprobar su capacidad diagnóstica.
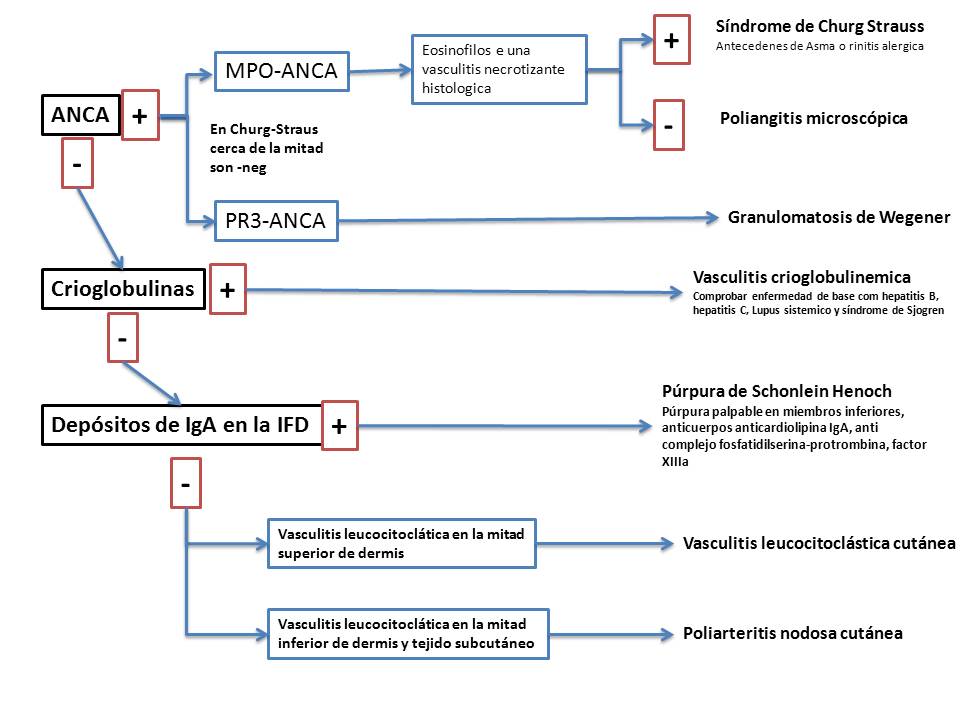{kind=link}
- Confirmar el diagnóstico de vasculitis mediante la realización de una biopsia cutánea para tener el diagnóstico definitivo de vasculitis, ver el tipo de infiltrado, el tamaño del vaso afecto y realizar estudios de inmunofluorescencia directa para demostrar la presencia de inmunocomplejos.
- Examinar la afectación de los órganos sistémicos para establecer si existe afectación cutánea o sistémica.
- Estudiar las posibles causas, intentado determinar si es una vasculitis primaria (dentro de los síndromes de vasculitis primarios) o secundaria y establecer el tratamiento.
Pasos a realizar ante una vasculitis cutánea

- Descartar una causa infecciosa : dado que el tratamiento de las vasculitis por infección directa de los vasos (vasculitis séptica) es completamente diferente de las vasculitis mediada inmunológicamente, esta causa debe ser descartada en el inicio de la valoración de un enfermo con vasculitis.
- Si se determina que es una vasculitis mediada inmunológicamente debe determinarse si es de tipo III, mediada por inmunocomplejos y deben buscarse el origen de estos inmunocomplejos, que puede ser exógeno (medicación, infección, proteínas) o exógeno (DNA, inmunoglobulinas, antígenos tumorales). La determinación de la existencia de la formación de inmunocomplejos como causa de la vasculitis es tranquilizadora ya que aumenta las posibilidades de que la vasculitis sea autolimitada y facilita la retirada de la fuente del antígeno (bien por retirada de la medicación o por tratamiento de la enfermedad de base).
- En relación a la clínica del paciente, se establecerá una serie de exploraciones para determinar la afectación sistémica. Los signos y síntomas que sugieren vasculitis en otros órganos
- Afectación muscular: Mialgias con elevación de enzimas musculares
- Afectación digestiva: Dolor abdominal con sangre oculta en feces o elevación de enzimas pancreáticos
- Afectación cardiaca: Angina con elevación de enzimas micocardicos
- Afectación renal: Hematuria con proteinuria
- Afectación de nervios periféricos: Mononeuritis múltiple con defectos en la conducción nerviosa
- Afectación de sistema nervioso central: Disfunción cerebral o visual
- Test serológicos: ANA, Crioglobulinas, Anticuerpos anti hepatitis B y C, ANCA, y Niveles de complemento.
Tratamiento de las vasculitis leucocitoclástica cutánea
En muchos pacientes la vasculitis de pequeño vaso va a tener un curso relativamente benigno, autolimitado, especialmente si la enfermedad está limitada a la piel. Sin embargo, los pacientes con enfermedad agresiva como los pacientes con vasculitis de pequeño vaso asociado a ANCAS necesita iniciar el tratamiento de forma rápida y agresiva.1-De soporte: reposo, elevación de las partes declives y protección frente a traumatismos y frío
2-Antiinflamatorios: corticoides tópicos, antiinflamatorios no esteroideos: Indometacina,
3-Antiagregantes plaquetarios: aspirina, dipiridamol.
4-Sistémicos: corticoides sistémicos y medicaciones inmunosupresoras.
casos per a discussió a classe
bibliografia
- ANCA associated vasculitis
- crioglobulinemias
- Gonzalez-Gay, MA, Garcia-Porrua C, Pujol RM. Clinical approach to cutaneous vasculitis. Curr Op Rheumatol, 2005, 17:56
- Carlson JA. The histological assessment of cutaneous vasculitis. Histopathology 2010 56:3-23
- Carlson JA; Chen KR. Cutaneous vasculitis update: small vessel neutrophilic vasculitis syndromes. Am J Dermatopath 2006, 28:486
- Kawakami T. New algorithm (kawakami algorithm) to diagnose primary cutaneous vasculitis. J dermatolo. 2010 37:113-124
- Bosch X; Guilabert A; Font J. Antineutrophil cytoplasmic antibodies. Lancet 2006; 368(9533):404-418.
- Jennette JC, Falk RJ. Small vessel vasculitis. N Eng J Med 1997, 337:1512
- Ledford DK. Immunologic aspects of vasculitis and cardiovascular diseases. JAMA 1997, 278:1962.
- Lotti T, y col. Cutaneous small-vessel vasculitis. J Am Acad Dermatol 1998, 39:667.
- Buerguer disease
- petechial rash on the extremities. cutaneous leucocitoclastic vasculitis
- Kawasaki disease
Suscribirse a:
Entradas (Atom)