1.- DefiniciónLa insuficiencia suprarrenal es la consecuencia de la disminución en la síntesis de glucocorticoides por parte de la glándula suprarrenal asociada o no con la disminución de mineralocorticoides. Esta falla puede ser causada por enfermedades que comprometen a la glándula suprarrenal (Primaria), a la glándula hipófisis y en consecuencia a la secreción de ACTH (Secundaria) o que comprometen al hipotálamo y a la secreción de CRH (Terciaria).
2.- Diagnóstico de Sospecha
La causa más frecuente de insuficiencia suprarrenal es la suspensión del tratamiento crónico con glucocorticoides. Los síntomas suelen ser larvados, ninguno de ellos es patognomónico y es importante un alto índice de sospecha por parte del médico para arribar al diagnóstico. La astenia y la fatigabilidad son muy frecuentes y precoces, tanto en la suspensión aguda de corticoides como en el Addison. La pérdida de peso es un síntoma cardinal del déficit crónico. Las causas son múltiples: anorexia, náuseas, vómitos y el síndrome de malabsorción asociado con autoinmunidad. Uno de los efectos más graves de la deficiencia crónica de glucocorticoides es la hipoglucemia que se presenta espontáneamente o inducida por la ingesta de alcohol. Los síntomas de neuroglucopenia son mayores que los esperables por el nivel de glucemia pero se acompañan de escasa expresión vasomotora periférica como consecuencia, al menos en parte, de la disminución de la secreción de adrenalina suprarrenal. La hipotensión ortostática, es característica en el déficit esteroideo crónico pudiendo llegar al síncope. La hipovolemia y la hiponatremia no son la única causa de la hipotensión ya que ésta se presenta aún con volemia normal y sin déficit del contenido corporal de sodio. La contractilidad miocárdica y el volumen minuto pueden estar disminuidos y son reversibles con la administración de glucocorticoides. La situación reviste más gravedad en la enfermedad de Addison que en el déficit glucocorticoideo secundario porque se suma la falta de mineralocorticoides los que tienen efecto propio sobre el miocardio aumentando el inotropismo.
En la insuficiencia suprarrenal aguda se produce hipovolemia con un rápido pasaje de sodio y de agua del espacio extracelular al intracelular con una marcada disminución del flujo plasmático renal y del filtrado glomerular que se normalizan con la administración de glucocorticoides. Tanto el hipopituitarismo como la insuficiencia suprarrenal primaria pueden causar hiponatremia; en el primer caso debido al déficit de cortisol que, su vez, puede dar lugar a una secreción inapropiada de hormona antidiurética más aún si está asociado con hipotiroidismo, en tanto que en la deficiencia primaria existe además una insuficiencia mineralocorticoide que contribuye adicionalmente a la hiponatremia y, eventualmente, a la hiperkalemia.
Etiología de la insuficiencia suprarrenal primariaLa etiología de la Enfermedad de Addison puede agruparse en 3 categorías: disgenéticas, por fallo en la esteroidogénesis y destructivas. Su frecuencia está fuertemente vinculada a la edad y al sexo de los pacientes.
-Disgenéticas: los defectos del desarrollo suprarrenal incluyen mutaciones en factores de transcripción como el factor-1 esteroidogénico (SF-1), del DAX-1y del gen del receptor de ACTH La hipoplasia como parte de síndromes como el de Smith-Lemli-Opitz, el síndrome triple A (acalasia, alacrimia, Addison), el síndrome de Kearns-Sayre causado por deleciones del DNA mitocondrial y en la deficiencia de glicerol quinasa. El síndrome familiar de resistencia a la ACTH (Síndrome de Allgrove) enfermedad autosómica recesiva caracterizada por un déficit de cortisol y andrógenos que no responden al estímulo con ACTH.
-Fallo en la esteroidogénesis: en los niños de ambos sexos por defectos enzimáticos de la 3 beta-hidroxiesteroide dehidrogenesa y de la 21 hidroxilasa. Esta última es la causa más común de insuficiencia suprarrenal aguda en las 2-3 primeras semanas de vida. La hiperplasia suprarrenal congénita lipoídica originada en una mutación en el gen StAR e insuficiencia suprarrenal relativa en pacientes con abetalipoproteinemia por ausencia de LDLs o del receptor de LDL, ya que a partir del colesterol que aportan las LDL y las HDL circulantes se sintetiza la mayor parte del cortisol.
-Enfermedades destructivas: la etiología autoinmune es la causa más frecuente, puede presentarse en forma aislada o como parte del síndrome poliglandular autoinmune (SPG) de tipo1 y 2.
El SPG tipo 1 es una rara enfermedad autosómica recesiva que afecta por igual a ambos sexos y se manifiesta clínicamente durante la infancia. Se caracteriza por 3 componentes principales: candidiasis mucocutánea, hipoparatiroidismo y enfermedad de Addison (anticuerpos antisuprarrenales positivos). Es frecuente también la presencia de hepatitis crónica, síndrome de mala absorción (enfermedad celíaca), alopecía universal.
El SPG tipo 2, por su parte, tiene como manifestaciones asociadas más comunes a la enfermedad de Addison y la tiroiditis autoinmune (síndrome de Schmidt). Es más frecuente en las mujeres y si bien puede presentarse a cualquier edad, el pico en la edad adulta, es alrededor de los 30 años. Frecuentemente se asocian gastritis atrófica (anemia perniciosa), vitiligo y diabetes tipo 1. Estas diversas enfermedades autoinmunes pueden presentarse en forma simultánea o sucesiva, lentamente a lo largo de años. Los autoanticuerpos anti 21 hidroxilasa se observan en el 70-80% de los pacientes con enfermedad de Addison. Al momento del diagnóstico la proporción de sueros positivos es más alta (95%) y declina alrededor del 50% después de los 20 años de evolución. En los niños con adrenalitis autoinmune los anticuerpos son positivos en el 100% de los casos.
La adrenoleucodistrofia es una enfermedad ligada al cromosoma X caracterizada por un defecto en la oxidación y acumulación de los ácidos grasos de cadena larga con insuficiencia suprarrenal y desmielinización progresiva del SNC.
En los países desarrollados entre un 12-20% de los pacientes tienen como causa a la tuberculosis, no existen estadísticas sobre prevalencia etiológica del Addison en nuestro medio. La insuficiencia suprarrenal puede presentarse tardíamente, 20 años después de la infección prima-ria, o durante el proceso agudo, por un extenso fenómeno destructivo de la glándula. Las micosis (blastomicosis e histoplasmosis) son muy infrecuentes.
La hemorragia de la glándula por anticoagulantes, una sepsis de gérmenes Gram negativos o en el síndrome antifosfolipidíco.
En pacientes con SIDA , la colonización suprarrenal por gérmenes oportunistas, por modificaciones en la estructura del receptor de ACTH suprarrenal o del receptor a glucocorticoides inducidas por el virus.
Diversas drogas pueden provocar insuficiencia suprarrenal: metopirona, Op'DDD y ketoconazol, que pueden utilizarse para el tratamiento del síndrome de Cushing, y otras que interfieren la degradación hepática del cortisol por inducción enzimática (rifampicina, anticonvulsivantes, mitotano, hipertiroxinemia) o por inhibición (antiretrovirales) por lo que será necesario ajustar la dosis de cortisol.
Etiología de la insuficiencia suprarrenal secundariaTodo proceso que destruya tejido hipofisario e interfiera con la secreción de la ACTH puede causar insuficiencia suprarrenal secundaria, entre ellas, grandes tumores hipofisarios, enfermedades infecciosas (TBC, histoplasmosis) o infiltrativas, hipofisitis linfocitarias, traumatismo craneano o grandes aneurismas intraselares, infarto hipofisario secundario a la hemorragia e hipotensión postparto (síndrome de Sheehan), hemorragia intratumoral (apoplejía hipofisaria) y las metástasis. El déficit de ACTH puede ser único o asociarse con otros déficits hormonales (panhipopituitarismo). La suspensión del tratamiento glucocorticoideo exógeno y la resección del tumor hiperfuncionante en el síndrome de Cushing causan insuficiencia secundaria y terciaria.
3.- Confirmación del diagnóstico
a.- ¿Basales? ¿De qué?
b.- ¿Test de ACTH? En aquellos pacientes que refieren el antecedente de la suspensión de corticoides exógenos o con enfermedades que potencialmente pueden comprometer al eje hipotálamo-hipófiso-suprarrenal, no existe teóricamente dificultad para el diagnóstico. El diagnóstico de insuficiencia suprarrenal debe sospecharse frente a un paciente hipotenso, hipoglucémico, con vómitos, malestar general, descenso de peso, confuso o en coma cuyo cuadro fue precipitado, por ejemplo, por una infección o una cirugía. El diagnóstico de la insuficiencia suprarrenal se con-firma demostrando un cortisol inapropiadamente bajo y la determinación de ACTH indicará si es primaria (elevado) o secundaria (normal o bajo). En los exámenes de rutina los hallazgos clásicos incluyen: hiponatremia (88%), hiperkalemia (64%), urea elevada, anemia, eosinofilia, linfocitosis e hipoglucemia. Sin embargo, en el momento actual en que la sospecha diagnóstica suele ser más precoz, algunos de estos signos pueden estar ausentes.
Fisiológicamente la concentración plasmática de cortisol es elevada a primeras horas de la mañana (4-8h), una concentración aislada menor de 3 μg/dl (30 ng/dl, 80 nmol/l) es fuertemente sugestiva de insuficiencia suprarrenal, valores menores de10 μg/dl (10 ng/dl, 275 nmol/l) son sospechosos, pero se requiere de una prueba de estímulo confirmatoria. Una concentración de cortisol matinal mayor de 15 -18 μg/dl predice una respuesta normal al estímulo en prácticamente todos los pacientes. No se recomienda la determinación basal del cortisol libre en saliva o urinario que pueden estar dentro del rango normal bajo.
Una concentración inapropiadamente baja de cortisol con ACTH plasmática elevada es sugestiva de insuficiencia suprarrenal primaria. Por otro lado, niveles de cortisol y ACTH matinal inapropiadamente bajos sugieren enfermedad secundaria o terciaria, en estos casos es conveniente realizar la RM selar para descartar el compromiso tumoral.
Una vez confirmada la insuficiencia suprarrenal prima-ria se confirmará la etiología autoinmune con los anticuerpos antiadrenales. Si estos son negativos, debe solicitarse la tomografía computada para avanzar en el diagnóstico etiológico. Un marcado agrandamiento suprarrenal bilateral con o sin calcificaciones es un hallazgo usual en TBC, micosis, amiloidosis, enfermedades granulomatosas y metástasis. En estos casos, se sugiere la punción con aguja fina guiada por tomografía.
Prueba de hipoglucemia insulínicaLa hipoglucemia causa una respuesta mayor de estrés provocando un aumento de ACTH, cortisol, GH, prolactina y activación del sistema nervioso simpático. La activación del eje hipotálamo-hipófiso-suprarrenal que ocurre durante la hipoglucemia insulínica es equivalente a la que se produce durante una cirugía mayor en pacientes sanos. A pesar de ser considerada la prueba "gold standard" para el diagnóstico de insuficiencia suprarrenal es potencialmente riesgosa y requiere control médico estricto. Esta prueba está contraindicada en ancianos, pacientes con cardiopatía, enfermedad cerebrovascular o antecedentes convulsivos.
Se considera una respuesta normal, un valor de cortisol superior a 18 μg/dl (180 ng/dl, 500 nmol/l). A pesar de ser considerada una prueba patrón se propone utilizarla sólo ante la sospecha de un déficit aislado de ACTH de comienzo reciente o asociado al déficit de hormona de crecimiento o cuando los otros estudios realizados no hayan sido concluyentes.
Prueba de estímulo con ACTHLa prueba de estímulo con análogo de corticotrofina 250 μg debe realizarse en todo paciente en que se sospeche insuficiencia suprarrenal, a menos que los estudios basales de cortisol y ACTH establezcan claramente el diagnóstico.
Para descartar insuficiencia suprarrenal secundaria a la cirugía hipofisaria, la prueba debe realizarse luego de 4 semanas del postoperatorio para evitar un falso negativo.
La sensibilidad para el diagnóstico de insuficiencia primaria es del 97,6% y para el fallo secundario del 57%.
La concentración de ACTH plasmática luego de inyectar 250 μg alcanza los 60.000 pg/ml y con la administración de 1 μg es de 1900 pg/ml semejante a la respuesta fisiológica al estrés endógeno y a la hipoglucemia insulínica. Es lógico asumir que una dosis de 250 μg pueda sobreestimular una glándula parcialmente atrofiada y producir una respuesta "normal". Esto dio lugar al uso de un test de baja dosis con 1 μg para el estudio del fallo suprarrenal secundario. Entre las desventajas de la prueba están la necesidad de diluir la ampolla de 250 μg, la variabilidad en el pico de cortisol y la falta de validación en pacientes agudamente enfermos. El diagnóstico es más difícil cuanto más leve sea la alteración del eje adrenal.
Se define como respuesta normal una concentración de cortisol superior a 18 μg /dl (180ng/dl, 500 nmol/l) sin considerar el incremento porcentual.
Insuficiencia corticotropa aislada versus corticoterapiaAnte un resultado que confirme insuficiencia suprarrenal, que cursa con ACTH baja o "normal", junto con RM selar normal, se plantean diversos diagnósticos, entre ellos corticoterapia, trauma o déficit aislado de ACTH.
La terapia crónica con glucocorticoides, es la causa más frecuente de insuficiencia suprarrenal, más del 50% de los pacientes que reciben por largo tiempo dosis moderadas o altas de glucocorticoides, pueden presentarla. Además del síndrome de Cushing típico, la corticoterapia crónica produce la supresión del eje hipotálamo hipófiso suprarrenal por la inhibición que se genera en la síntesis y liberación de CRH y ACTH.
El tiempo en que se desarrolla la supresión y la recuperación dependen de la dosis, del tipo de glucocorticoide, del tiempo y de la vía de administración. Si bien dosis suprafisiológicas por más de 4 semanas suprimen el eje hipotálamo-hipófiso-suprarrenal, dosis menores y por tiempos más cortos también pueden afectarlo.
La recuperación en tiempo es variable, se puede diferenciar en etapas, no siempre claramente delimitadas, inicialmente comienza a subir ACTH, luego se normaliza el cortisol, y por último se normaliza la respuesta a ACTH exógena. El tiempo de recuperación es muy variable, des-de semanas hasta los 18 meses.
Dentro de las causas poco frecuentes de déficit de ACTH podemos mencionar: - Causas genéticas con déficit plurihormonal: mutaciones del PROP-1, de Pit -1 o factores de transcripción involucrados en desarrollo hipofisario. Con déficit aislado de ACTH: mutaciones génicas de la proopiomelanocortina (POMC), mutaciones de PC 1(prohormona convertasa 1) y mutaciones del factor de transcripción involucrado en la diferenciación del corticotropo.
-Causas autoinmunes: infiltración linfocitaria, anticuerpos anticorticotropos asociado o no a otras enfermedades autoinmunes.
- Otras: postraumática, postradioterapia cerebral, alcoholismo
8.- Tratamiento: a.- Glucocorticoides: Tipo y dosis. b.- Mineralocorticoides . c.- DHEA.
La hidrocortisona es el glucocorticoide de elección en el tratamiento de la insuficiencia suprarrenal. Los corticoides sintéticos (prednisolona, dexametasona) que se administran en una única toma, pueden utilizarse como alternativa en pacientes con poca adhesión al tratamiento; sin embargo se asocian a mayores efectos secundarios en el metabolismo óseo e hidrocarbonado.
Históricamente se calculó una producción diaria de cortisol de 12 a 15 mg/m2/día lo que llevó a un régimen de reemplazo con 30 mg de hidrocortisona. Estudios recientes indican que la producción diaria de cortisol sería de 5 a 10 mg/m2/día, lo que equivale a 15 a 25 mg/día de hidrocortisona de reemplazo. Aún pequeños excesos en el reemplazo glucocorticoideo parecen ocasionar un impacto cardiovascular adverso. Los pacientes que reciben 30 mg presentan mayor nivel de colesterol total, de triglicéridos, del índice de masa corporal y de la HbA1c comparados con aquellos tratados con 20 mg.
Para intentar remedar el ritmo circadiano normal, se aconseja repartir la dosis en 2 ó 3 tomas con la mitad o 2/3 por la mañana.
La hidrocortisona tiene una rápida absorción que lleva a alcanzar picos séricos suprafisiológicos de cortisol. Su vida media plasmática es de 1.7 horas y la vida media biológica de 6 a 10 horas. Esto hace que se encuentren valores muy bajos de cortisol en ayunas y antes de la ingesta del comprimido de hidrocortisona de la tarde.
Recientemente, se han presentado formulaciones de hidrocortisona de liberación prolongada que remedarían con mayor exactitud el ritmo circadiano. Todavía faltan ensayos clínicos controlados que demuestren la superioridad de estas nuevas formulaciones sobre los tratamientos convencionales.
A diferencia del hipotiroidismo primario, no existe en la insuficiencia suprarrenal un parámetro fiable y universalmente aceptado para monitorear el tratamiento. Para establecer si un paciente se encuentra sobre o subreemplazado se han utilizado mediciones seriadas de cortisol sérico a lo largo del día, cortisol urinario de 24 h, dosaje de ACTH y cortisol salival.
En la insuficiencia primaria, los niveles de ACTH matinales se encuentran frecuentemente elevados y disminuyen rápidamente luego de la primera dosis del día de hidrocortisona. No se debe tomar como meta la normalización de los niveles matinales de ACTH ya que esto llevaría invariablemente a un exceso en el reemplazo glucocorticoideo. La medición de cortisol libre urinario de 24 h también ofrece dificultades. Luego de la ingesta de la hidrocortisona se alcanzan niveles suprafisiológicos de cortisol y se satura la proteína trasportadora de corticoides (CBG) lo que lleva a una mayor excreción de cortisol urinario. Los niveles de cortisol plasmático y urinario luego de la administración de glucocorticoides muestran una marcada variabilidad individual.
La medición seriada de cortisol plasmático a lo largo del día no ha sido validada con estudios controlados como parámetro objetivo de seguimiento.
El monitoreo del tratamiento se basa principalmente en parámetros clínicos controlando periódicamente peso, tensión arterial, perímetro de cintura y detectar posibles síntomas de exceso o déficit del reemplazo glucocorticoideo. A pesar del tratamiento, los pacientes con insuficiencia suprarrenal presentan una mayor mortalidad que la población general, deterioro de la calidad de vida, mayor riesgo de sufrir eventos vasculares y alteraciones del metabolismo fosfocálcico.
En la insuficiencia suprarrenal autoinmune, se debe monitorear la aparición de patologías asociadas: vitiligo, anemia perniciosa, disfunción tiroidea, diabetes mellitus y celiaquía.
En pacientes con hipotiroidismo e insuficiencia suprarrenal se deben administrar primero los glucocorticoides que las hormonas tiroideas para evitar que se desencadene una crisis suprarrenal por el aumento de la metabolización hepática del cortisol.
En la insuficiencia suprarrenal primaria el reemplazo con mineralocorticoides se realiza con 9α-fludrocortisona.
La dosis puede variar entre 0.05 y 0.25 mg/día, en una toma matinal para mantener la renina plasmática en el límite superior de lo normal. Clínicamente se debe controlar la presencia de edemas, la presión arterial de reposo e hipotensión ortostática.
La DHEA no es un tratamiento universalmente aceptado, se reserva para pacientes con menor calidad de vida a pesar del reemplazo glucocorticoideo adecuado especial-mente en mujeres con síntomas de déficit androgénico. La dosis de reemplazo es de 25 a 50 mg, una vez por día, en la mañana.
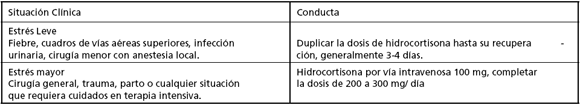
9.- Pautas de estrés. Medidas generales. Adecuación de la dosisUna parte crucial en el seguimiento de la insuficiencia suprarrenal crónica es la educación al paciente y su entorno para enfrentar situaciones de emergencia. El paciente debe portar un brazalete o collar que acredite su condición de insuficiente suprarrenal con instrucciones claras y concisas Las pautas de estrés deben hacerse extensivas a los pacientes con tratamiento corticoideo crónico. Los pacientes deben disponer en su domicilio de una preparación parenteral de glucocorticoides (hidrocortisona o dexametasona de vida media más prolongada, etc.) para ser utilizada en caso de emergencia o de intolerancia digestiva (vómitos y/o diarrea). Tras la administración del corticoide endovenoso el paciente debe consultar de inmediato al médico para el tratamiento específico, de la enfermedad que lo descompensó. Se administrará el esquema glucocorticoideo de estrés en tanto se resuelve la enfermedad que indujo a la descompensación, yugulada ésta se regresará al tratamiento habitual.
Etiología de la Insuficiencia SuprarrenalInsuficiencia Suprarrenal Primaria
· Autoinmune (Síndrome Poliglandular)
· Infecciosas:Tuberculosisy Micosis (histoplasmosis,blastomicosis)
· Sarcoidosis, amiloidosis, hemocromatosis
· Hemorragia (meningococcemia, anticoagulantes,traumatismo)
· Infiltración neoplásica (riñón) o metástasis (riñón, melanoma, mama, pulmón)
· Hiperplasia Suprarrenal Congénita Clásica
· Hipoplasia/Disgenesia Suprarrenal Congénita
· Síndrome de Resistencia al ACTH
· Adrenoleucodistrofia
· Síndrome de Inmunodeficiencia Adquirida
· Suprarrenalectomía bilateral
· Inhibidores de la síntesis de esteroides (metopirona, ketoconazol, aminoglutetimida)
· Drogas adrenolíticas (o,p'DDD)
· Antagonistas de los glucocorticoides (RU 486)
Insuficiencia Suprarrenal Secundaria
· Suspensión de glucocorticoides exógenos o de ACTH
· Postoperatorio del tumor suprarrenal secretante de cortisol
· Postoperatorio del tumor hipofisario secretante de ACTH
· Lesiones hipofisarias o hipotalámicas:
Tumores
Enfermedades inflamatorias
Infecciones
Enfermedades autoinmunes
Infilitración granulomatosa
Traumatismo cráneo encefálico
Aplasia, hipoplasia, displasia o ectopía congénitas
Cirugía hipotálamo hipofisaria
Radioterapia hipotálamo hipofisaria
Hemorragia hipotálamo hipofisaria (apoplejía)
Déficit aislado de ACTH
· Tumores Enfermedades inflamatorias Infecciones Enfermedades autoinmunes Infilitración granulomatosa Traumatismo cráneo encefálico Aplasia, hipoplasia, displasia o ectopía congénitas Cirugía hipotálamo hipofisaria Radioterapia hipotálamo hipofisaria Hemorragia hipotálamo hipofisaria (apoplejía) Déficit aislado de ACTH
• Deficiencia familiar de CBG
Elementos Clínicos Sugestivos de Insuficiencia Suprarrenal
• Hipoglucemia inexplicable
• Pérdida de peso
• Desproporción entre la severidad de la enfermedad actual y el grado de postración por hipotensión arterial, colapso circulatorio y deshidratación
• Hallazgos endocrinos: pérdida del vello pubiano, esterilidad, hipotiroidismo
• Vitiligo
• Hiperpigmentación de piel ( zonas de roce, pliegues, cicatrices) y mucosas
• Debilidad general y depresión sin síndrome psiquiátrico evidente
• Síntomas gastrointestinales (vómitos, epigastralgia)
• Shock refractario a drogas vasoactivas
Tratamiento de la Insuficiencia Suprarrenal Crónica
• Glucocorticoides . Primaria: Hidrocortisona 20 a 25 mg/día
• Secundaria: Hidrocortisona 15 a 20 mg/día
• Dividido en dos o tres dosis
• Mineralocorticoides Sólo en insuficiencia primaria
• Fludrocortisona 0.05-0.2 mg/día
• Andrógenos suprarrenales Cuestionado, se beneficiarían mujeres con síntomas de déficit androgénico
• DHEA 25-50 mg/día
Seguimiento del paciente con insuficiencia adrenal:• No solicitar ACTH para ajustar la dosis de hidrocortisona
• CLU de 24 h, valor relativo
• Cortisol seriado, valor relativo
• Guiarse por los parámetros clínicos
• Mantener la Actividad de Renina Plasmática en el límite superior normal
• Disminuir la dosis de hidrocortisona en pacientes que reciben 30mg/día
• Mostrar al paciente beneficios del ajuste de dosis (peso, TA, glucemia)
• Educación para la prevención de la crisis
• Buscar patologías autoinmunes asociadas
• Reevaluación diagnóstica en casos dudosos